Analýza genomických a proteomických dat |
Analýza sekvencí DNA |
Sekvence |
Genom |
Sekvenování genomu |
Polymerázová řetězcová reakce |
Celogenomové sekvenování |
Genetické databáze |
Pyrosekvenování |
Sekvenování pomocí syntézy |
Iontové polovodičové sekvenování |
Sekvenování jedné molekuly DNA |
Sangerovo sekvenování |
Sestavení sekvence |
Vyhledávání v databázích |
Sekvence v GenBance |
Stahování sekvencí |
Informační zdroje pro proteiny |
BLAST-Vyhledávání podobných sekvencí |
Základní informace |
Výstupy z výukové jednotky |
Využití blastu |
Přístup k blastu |
Princip blastu |
E-hodnota |
Programy blastu |
Predikce genů a anotace sekvence DNA |
Nukleotidový blast |
Proteínový blast |
Blast využívající překlad DNA do sekvence aminokyselin a opačně |
Prohledávání specifických databází |
Vícenásobné vyhledávání |
Výsledek a interpretace |
Taxonomie nebo fylogeneze nalezených záznamů |
Základní informace |
Výstupy z výukové jednotky |
Komparativní anotace |
Anotace ab initio - od začátku |
Alignment |
Genomické ostrovy |
Otevřený čtecí rámec |
Predikce eukaryotických genů |
Skrytý markovův model |
Modelování začátku intronu |
Modelování frekvence kodonů |
Predikce jiných RNA molekul |
Příprava sekvence do genetických databází |
Základní informace |
Výstupy z výukové jednotky |
Lokální alignment |
Globální alignment |
Vícenásobný alignment |
Praktické problémy s alignmentem a jejich řešení |
Modelování příbuznosti sekvencí DNA |
Substituční model |
Substituce |
Genetické vzdálenosti |
Parametry substitučního modelu |
Příklady nejběžnějších substitučních modelů |
Heterogenita rychlosti evoluce mezi pozicemi |
Výběr substitučního modelu |
Metoda nejbližšího souseda |
Hierarchický test poměru věrohodností |
Akaikovo informační kriterium |
Bayesovo informační kritérium |
Inserce a delece |
Základní informace |
Výstupy z výukové jednotky |
Vstupní údaje pro metodu nejbližšího souseda |
Algoritmus NJ shlukování |
Výhody a nevýhody NJ metody |
Využití NJ metody |
Ověření stability uzlů stromu – bootstrap |
Fylogenetika |
Čtení fylogenetického stromu |
Fylogenetický strom formálně |
Fylogenetický strom intuitivně |
Počet možných stromů |
Vlastnosti fylogeneze |
Ancestrální sekvence |
Určení kořene stromu |
Využití fylogenetické informace |
Maximální věrohodnost |
Bayesiánská inference |
Markovovy řetězce Monte Carlo |
Efektivita prohledávání krajiny stromů |
Priory |
Autokorelace MCMC |
Burnin |
Posterior |
Diagnostika konvergence |
Divergence druhů |
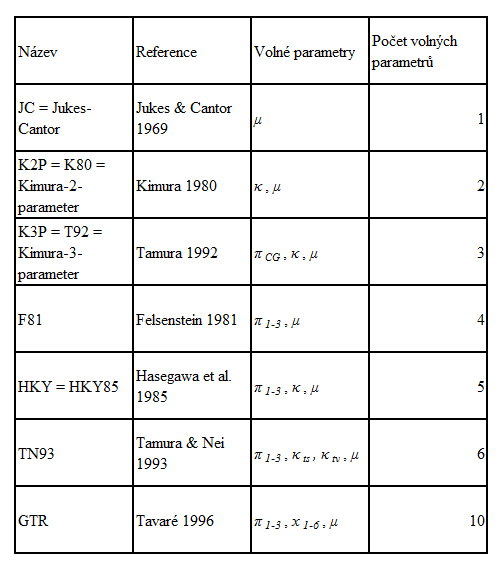
Příklady nejběžnějších substitučních modelů
Model popsaný výše se nazývá obecný časově-reverzibilní model (general time-reverzible model, GTR). Naopak, nejjednodušším modelem je model Jukese a Cantora (Jukes-Cantor model, JC; Tab.2). Modely HKY a GTR se používají nejčastěji, přičemž zvyšující se délka studovaných sekvencí umožňuje častěji správně a přesně stanovit parametry GTR modelu. Ten zároveň nejpřesněji modeluje evoluci sekvence DNA.
Tab. 2: Základní substituční modely a jejich charakteristika. Pokud není uvedeno jinak, modely předpokládají stejné frekvence nukleotidových bází a stejné rychlosti substitucí.