Analýza genomických a proteomických dat |
Analýza sekvencí DNA |
Sekvence |
Genom |
Sekvenování genomu |
Polymerázová řetězcová reakce |
Celogenomové sekvenování |
Genetické databáze |
Pyrosekvenování |
Sekvenování pomocí syntézy |
Iontové polovodičové sekvenování |
Sekvenování jedné molekuly DNA |
Sangerovo sekvenování |
Sestavení sekvence |
Vyhledávání v databázích |
Sekvence v GenBance |
Stahování sekvencí |
Informační zdroje pro proteiny |
BLAST-Vyhledávání podobných sekvencí |
Základní informace |
Výstupy z výukové jednotky |
Využití blastu |
Přístup k blastu |
Princip blastu |
E-hodnota |
Programy blastu |
Predikce genů a anotace sekvence DNA |
Nukleotidový blast |
Proteínový blast |
Blast využívající překlad DNA do sekvence aminokyselin a opačně |
Prohledávání specifických databází |
Vícenásobné vyhledávání |
Výsledek a interpretace |
Taxonomie nebo fylogeneze nalezených záznamů |
Základní informace |
Výstupy z výukové jednotky |
Komparativní anotace |
Anotace ab initio - od začátku |
Alignment |
Genomické ostrovy |
Otevřený čtecí rámec |
Predikce eukaryotických genů |
Skrytý markovův model |
Modelování začátku intronu |
Modelování frekvence kodonů |
Predikce jiných RNA molekul |
Příprava sekvence do genetických databází |
Základní informace |
Výstupy z výukové jednotky |
Lokální alignment |
Globální alignment |
Vícenásobný alignment |
Praktické problémy s alignmentem a jejich řešení |
Modelování příbuznosti sekvencí DNA |
Substituční model |
Substituce |
Genetické vzdálenosti |
Parametry substitučního modelu |
Příklady nejběžnějších substitučních modelů |
Heterogenita rychlosti evoluce mezi pozicemi |
Výběr substitučního modelu |
Metoda nejbližšího souseda |
Hierarchický test poměru věrohodností |
Akaikovo informační kriterium |
Bayesovo informační kritérium |
Inserce a delece |
Základní informace |
Výstupy z výukové jednotky |
Vstupní údaje pro metodu nejbližšího souseda |
Algoritmus NJ shlukování |
Výhody a nevýhody NJ metody |
Využití NJ metody |
Ověření stability uzlů stromu – bootstrap |
Fylogenetika |
Čtení fylogenetického stromu |
Fylogenetický strom formálně |
Fylogenetický strom intuitivně |
Počet možných stromů |
Vlastnosti fylogeneze |
Ancestrální sekvence |
Určení kořene stromu |
Využití fylogenetické informace |
Maximální věrohodnost |
Bayesiánská inference |
Markovovy řetězce Monte Carlo |
Efektivita prohledávání krajiny stromů |
Priory |
Autokorelace MCMC |
Burnin |
Posterior |
Diagnostika konvergence |
Divergence druhů |
Sangerovo sekvenování
Sangerovo sekvenování umožňuje v každé reakci přečíst jenom jeden namnožený úsek DNA bez masivní paralelizace. Sekvenační PCR probíhá za přítomnosti jednoho sekvenačního primeru a směsi deoxyribonukleotidů a fluorescenčně značených dideoxyribonukleotidů. Pokud se do syntetizovaného řetězce přiřadí dideoxyribonukleotid, PCR se zastaví. Výsledkem je směs různě dlouhých PCR produktů, které mají na konci fluorescenčně značený poslední připojený nukleotid. Tato směs se rozdělí podle délky pomocí kapilárové elektroforézy a laserem se přečte fluorescenční signál (obr. 1).
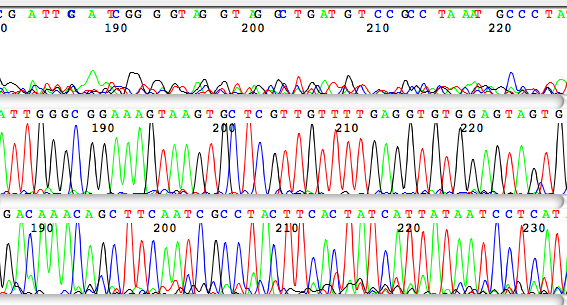
Obr. 1: Chromatogramy ze Sangerova sekvenování různé kvality. Horní sekvence je sice strojově přečtena ale signál je slabý, konfliktní a sekvence bude nesprávná. Prostřední chromatogram má jasně čitelnou sekvenci optimální kvality. Spodní chromatogram je akceptovatelný s opatrností, jelikož má téměř v celé délce rušivý signál.
