Otevřený čtecí rámec
Signál v sekvenci je nutný pro úspěšnou predikci genů prokaryot a eukaryot, ale využití přímo při hledání kandidátních úseků DNA má hlavně u prokaryotických genomů. Prokaryotické organizmy mají totiž geny blízko u sebe, někdy se dokonce i překrývají (Obr. 1). Jejich regulace bývá zjednodušená, kdy je několik genů sdružených do jednoho operonu se společnou regulací jedním promoterem. Predikce genů u prokaryot proto vychází z vyhledávání otevřených čtecích rámců (open reading frame – ORF; Obr.2). Jsou to úseky na jednom z řetězců DNA, které začínají start kodonem a končí stop kodonem.
Výjimka: Aminokyselina selenocystein je kódována kodonem TGA, což je běžně v genetickém kódu stop kodon. Změna kódování je signalizována sekvencí přímo navazující na kodon TGA, který by měl kódovat selenocystein. Některé nové algoritmy na predikci genů v současnosti začínají rozlišovat i selenoproteiny. V minulosti se ale selenoproteiny nepredikovaly.
ORF je nutné vyhledávat pro správně zvolenou translační tabulku. Genetický kód se totiž u některých skupin organizmů anebo u cytoplazmatických genomů (mitochondriální, chloroplatostový) částečně odlišuje od standartního genetického kódu.
Otázka: Kolik otevřených čtecích rámců je v sekvenci genomu RNA virů?
Tři, protože molekula RNA je jednořetězcová.
Otázka: Jaká je teoretická pravděpodobnost výskytu start kodonu v DNA sekvenci, která má obsah GC ? Předpokládejte, že nukleotidové báze se v sekvenci vyskytují na sobě nezávisle.
Guanin je komplementární k cytozinu, a proto v dvoušroubovici DNA s GC obsahem 40% bude guaninu i cytozinu po . Obdobně, všechny bázové páry, které v DNA nejsou GC jsou AT a jejich obsah je tedy 60%; adeninu a tyminu po 30%. Start kodon se na libovolném řetězci DNA bude vyskytovat s frekvencí:
Ne všechny start a stop kodony v sekvenci DNA prokaryot představují začátek nebo konec čtení kódující sekvence. Zúžení počtu kandidátních ORF probíhá na základě dodatečných informací: očekávané délky ORF (průměrná délka prokaryotického genu je asi 1kb) a výskytu specifických sekvencí v promoteru (Pribnowova sekvence: TATAAT v pozici 10bp a -35 sekvence TTGACA 35bp před začátkem transkripce. Pozor, počátek transkripce se vyskytuje vždy dřív než počátek translace - start kodon). V transkribované části genu se vyskytuje Shine-Dalgarnova sekvence, která má konsenzuální pořadí nukleotidů AGGAGG a představuje místo, kterým se mRNA navazuje na ribozom asi 8bp před start kodonem.
Jak je v biologických systémech obvyklé, uvedené sekvence se v genech téměř nikdy nevyskytují přesně. Mutace přímo v signálních sekvencích anebo v jejich vzdálenosti od start kodonu jsou běžné. Pro predikci genů z toho vyplývá, že charakterizace kontextu pro ORF je založena na pravděpodobnosti výskytu určitých nukleotidových bází v okolí start kodonu spíš než na vyhledání konkrétní sekvence.
Jednoduché pravidlo pro uživatele na kontrolu predikovaného genu je blast. Predikovaný gen se přeloží do sekvence aminokyselin a pomocí proteinového blastu se porovná se záznamy z genetických databází. K výsledkům je ale nutno přistupovat kriticky. Někdy se v databázích vyskytují anotace predikovaných genů jenom u několika organizmů. Může se jednat o chybné anotace, které se opakují při nezávislých predikcích a autoři je do databáze vložili bez ověření. Takové sekvence by vždy měly nést označení míry nejistoty anotace. Je na uživateli, zda u své sekvence podobnou anotaci uvede, nebo raději zvolí konzervativní přístup a anotuje jenom spolehlivě ověřitelné predikované geny.
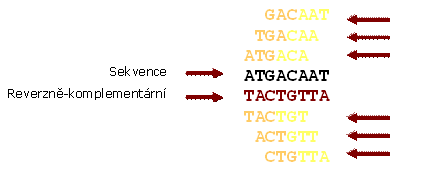
Obr. 2: Každá sekvence DNA má šest otevřených čtecích rámců podle pozice, na které začíná překlad sekvence nukleotidů v kodonech do sekvence aminokyselin.
