Praktický příklad analýzy
V tomto příkladu budeme podobně jako v předcházejících příkladech pracovat v softvéru R - pro statistické spracování dat. Pro obeznámaní s tímto programem doporučujeme pročíst si výukový text Analýza dat v R. V dalším textu budeme předpokládat, že program R znáte. Všechny příkazy níže se zadávají v do příkazového řádku v R konzole.
Budeme pokračovat v práci se stejným příkladem experimentu porovnávajícího ER (estrogen receptor) pozitivní a ER negativní karcinomy prsu, který jsme používali v předcházejících kapitolách. Budeme používat výsledky z praktického příkladu analýzy z kapitoly "Porovnávání skupin" a to zejména:
- objekt top100 obsahující top 100 odlišně exprimovaných genů mezi skupinami ER+ a ER- (jak získat top100 najdete ZDE)
- matici expresí X všech nádorů prsu (jak načíst tuto matici najdete ZDE)
a) Nejdříve vytvoříme heatmapu z top 100 odlišně exprimovaných genů mezi ER+ a ER-
Vyextrahujeme matici expresních dat o top100 genech, všechny vzorky:
> Y <- X[,as.character(top100)]
> dim(Y)
[1] 278 100
Zadefinujeme si barvy v heatmapě:
> barvy <- colorRampPalette(c("green", "black", "red"))(15)
Vykreslíme heatmapu:
> heatmap(X[,as.character(top100)], col=barvy, RowSideColors=as.character(as.numeric(clinical$ER_status)),scale="column")
> heatmap(Y, col=barvy, RowSideColors=as.character(as.numeric(clinical$ER_status)),scale="column")
.png)
Úloha k procvičení: vytvořte podobnou heatmapu pro top100 genů u skupin HER2+ vs HER2-.
b) Pokud přeneseme top100 genů z naší analýzy do nového datového souboru, dostaneme podobné shlukování? (Jinak řečeno validujeme naše pozorování?)
Načteme další datový soubor (TransBig) spolu s tabulkou klinických dat. Data lze stáhnout ZDE (nutno rozbalit archiv).
> load(file="TransBIG/TransBIG-expression.rdata")
> ls()
[1] "a" "adj_p_hodnoty" "b" "clinical" "clinical2"
[6] "cont.matrix" "d" "data.sam" "de" "de.adj"
[11] "de.adj.fin" "delta.table" "design" "de1" "ER.HER2spec"
[16] "ER.sam" "ERspec" "ER_vysledky" "ER_vysl_ord" "ER_vyzn"
[21] "ER_vyzn_DN" "ER_vyzn_UP" "farby" "fit" "fit2"
[26] "limma.res" "p_hodnoty" "samr.obj" "siggenes.table" "top100"
[31] "t_statistiky" "v" "X" "X2" "Y"
> clinical2 <- read.csv("TransBIG/TransBIG-Sample_info.csv")
> head(clinical2)
Patient.ID ER.pos.1.yes. Grade Lymph.node.invasion..1.yes. Age Tumor.size..mm.
1 4000471 0 3 0 57 30
2 4000473 1 2 0 46 35
3 4000475 1 3 0 51 40
4 4000477 1 NA 0 51 20
5 4000480 1 3 0 57 30
6 4000482 0 3 0 59 25
Time.RFS..days. Event.RFS
1 730 1
2 4931 1
3 511 1
4 7159 0
5 183 1
6 548 1
> ?load
> X2=X
Znovu načteme původní datovou matici:
> load(file="MDACC/GSE20194_MDACC_Expression.rdata")
> dim(X2)
[1] 253 1052
> X2_top100 <- X2[,intersect(colnames(X2), top100)]
> X2[1:5,1:5]
ACADS SPBC25 ADM PTPLB KIF21A
4000471 -2.042 0.054 5.639 1.474 1.069
4000473 -0.117 -0.609 -1.858 1.966 2.650
4000475 0.840 -2.033 -4.287 0.305 0.467
4000477 0.547 -1.088 -1.125 0.468 0.055
4000480 0.745 3.917 -3.891 0.097 1.952
> barvy <- colorRampPalette(c("green", "black", "red"))(15)
Vykreslíme druhou heatmapu jen s 9 geny, které jsme v druhé matici našli.
Přidáme parametr RowSideColors:
> heatmap(X2[,intersect(colnames(X2),top100)], col=barvy, RowSideColors=as.character(clinical2$ER.pos.1.yes))
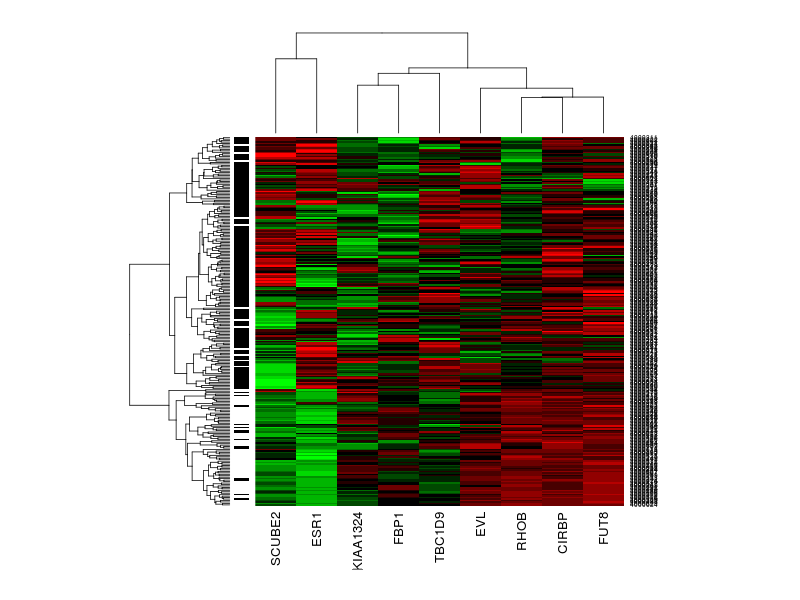
Porovname s původní heatmapou pokud máme jen 9 genů.
> windows();heatmap(X[,intersect(colnames(X2),top100)], col=farby, RowSideColors=as.character(as.numeric(clinical$ER_status)))
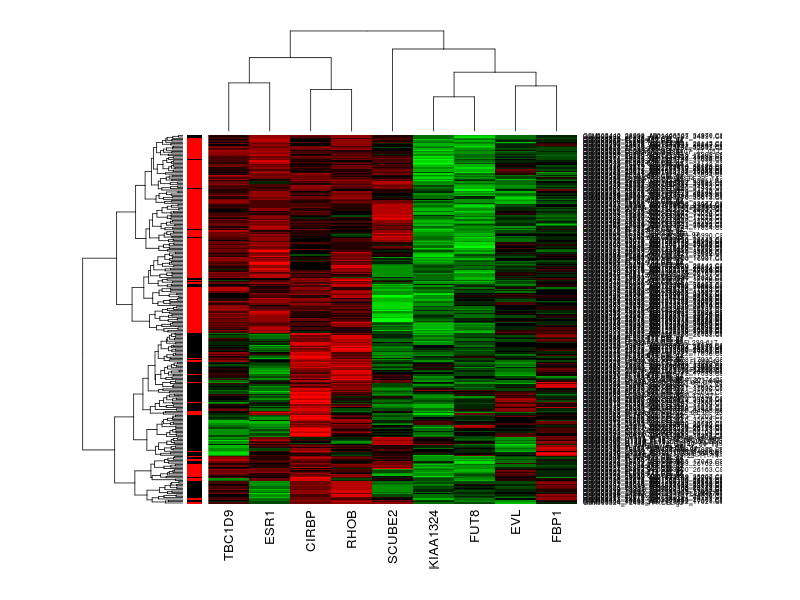
Již vizuálně vidíme, že geny, i když jich v novém souboru bylo pouze 9 ze 100, dokázalo rozdělit nádory prsu na ER+ a ER-.
