Kontrola kvality
Po načtení všech potřebných souborů provedeme základní kontrolu kvality dat a vypočteme základní statistiky.
1. Kontrola kvality dat: Zjistíme, jaké proměnné máme v souboru, ...
> names(clinical)
[1] "Sample.name" "title" "CEL.file" "source.name"
[5] "organism" "Age" "Race" "ER_status"
[9] "pCR_vs_RD" "PR_status" "molecule" "label"
[13] "description" "platform" "Additional.information" "Tbefore"
[17] "Nbefore" "BMNgrd" "ER" "HER2.Status"
[21] "Her2.IHC" "Her2.FISH" "Histology" "Treatment.Code"
[25] "Treatments.Comments"
..., jaké jsou jejich hodnoty.
> str(clinical)
'data.frame': 278 obs. of 25 variables:
$ Sample.name : Factor w/ 278 levels "FL1141-801(2)",..: 117 118 119 120 121 122 123 124 125 126 ...
$ title : Factor w/ 278 levels "BR_FNA_ML20",..: 27 42 32 49 6 24 31 48 23 53 ...
$ CEL.file : Factor w/ 278 levels "FL1141-801(2).CEL",..: 117 118 119 120 121 122 123 124 125 126 ...
$ source.name : Factor w/ 248 levels "Sample ID -- L20, fine-needle aspiration, breast cancer cells",..: 46 61 51 68 25 43 50 67 42 72 ...
$ organism : Factor w/ 1 level "homo sapiens": 1 1 1 1 1 1 1 1 1 1 ...
$ Age : int 57 69 77 54 75 29 50 42 61 38 ...
$ Race : Factor w/ 6 levels "","asian","black",..: 6 2 5 6 3 6 3 2 6 6 ...
$ ER_status : Factor w/ 2 levels "N","P": 2 2 2 2 1 1 2 1 1 2 ...
$ pCR_vs_RD : Factor w/ 2 levels "pCR","RD": 2 2 2 2 2 2 2 1 1 2 ...
$ PR_status : Factor w/ 2 levels "N","P": 2 2 1 1 1 1 1 1 2 1 ...
$ molecule : Factor w/ 1 level "total RNA": 1 1 1 1 1 1 1 1 1 1 ...
$ label : Factor w/ 1 level "biotin": 1 1 1 1 1 1 1 1 1 1 ...
$ description : Factor w/ 4 levels "MAQC_Distribution_Status: MAQC_Q -- Not used",..: 2 2 2 2 2 2 2 2 2 2 ...
$ platform : Factor w/ 1 level "GPL96": 1 1 1 1 1 1 1 1 1 1 ...
$ Additional.information: logi NA NA NA NA NA NA ...
$ Tbefore : int 2 2 4 2 2 4 2 2 1 3 ...
$ Nbefore : int 0 1 1 1 0 2 1 1 1 1 ...
$ BMNgrd : int 2 2 2 2 3 3 2 3 3 3 ...
$ ER : int 90 90 10 80 0 0 90 0 0 70 ...
$ HER2.Status : Factor w/ 2 levels "N","P": 1 1 2 1 1 1 1 2 1 1 ...
$ Her2.IHC : Factor w/ 9 levels "","ND","NEG",..: 7 4 7 NA 4 4 4 NA 4 4 ...
$ Her2.FISH : Factor w/ 102 levels "","ND","0","0.54",..: 70 70 79 44 70 70 70 72 70 70 ...
$ Histology : Factor w/ 18 levels "","IC/DCIS","IDC",..: 3 18 3 3 3 8 6 3 3 3 ...
$ Treatment.Code : Factor w/ 13 levels "","FAC","FACT",..: 7 7 7 7 7 12 7 7 7 7 ...
$ Treatments.Comments : Factor w/ 136 levels "","FAC x 3 (no response) Taxol x 8 (no response)",..: 132 93 94 107 121 92 85 86 126 106 ...
Factor je faktor, kategorická proměnná, nominální.
int je spojitá proměnná, integer
logi je logická proměnná, (TRUE, FALSE)
Je důležité identifikovat význam proměnných, které neznáme.
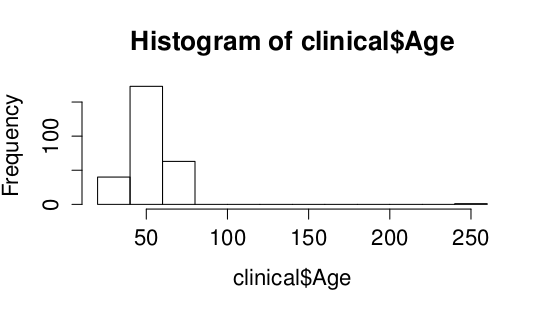
Pro kontrolu věku nejprve vykreslíme histogram:
> hist(clinical$Age)
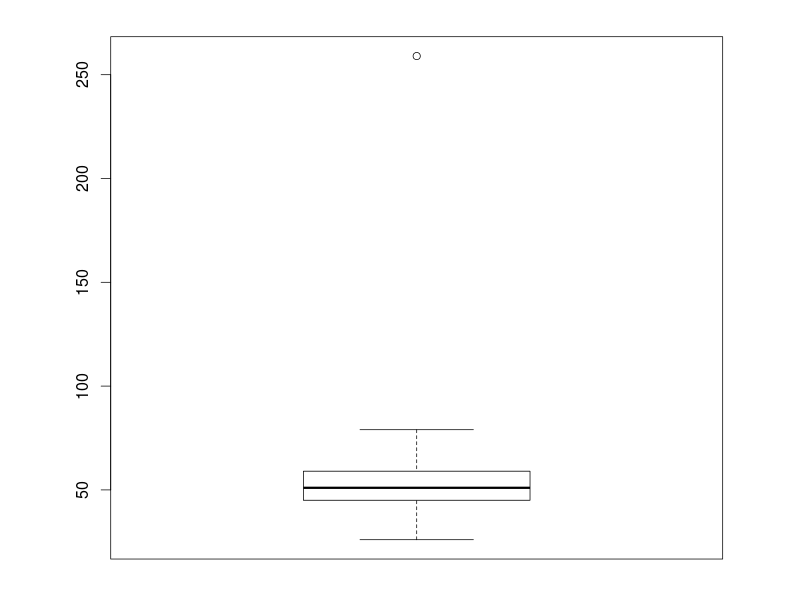
také boxplot:
> boxplot(clinical$Age)
na závěr vypočítáme statistiky:
> quantile(clinical$Age)
Error in quantile.default(clinical$Age) :
missing values and NaN's not allowed if 'na.rm' is FALSE
vzhledem k tomu, že máme v datech chybějící hodnoty, musíme zadat parametr, který zajistí, že chybějící hodnoty budou odstraněny z výpočtu:
> quantile(clinical$Age, na.rm=TRUE)
0% 25% 50% 75% 100%
26 45 51 59 259
Jsou naše výsledky v pořádku? Věk pacienta 259 je dost nepravděpodobný, zřejmě došlo k chybě při zadávání dat do počítače.
Nyní provedeme kontrolu kategoriální proměnné rasa:
Vykreslíme barplot:
> plot(clinical$Race)
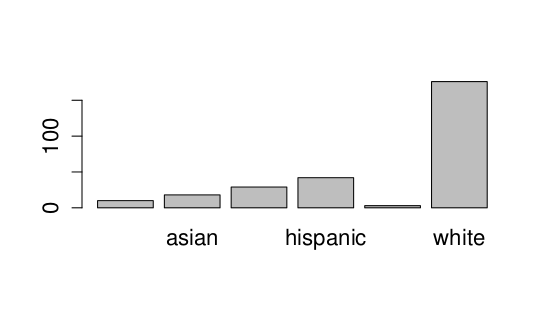
Jaká rasa je v souboru zastoupena nejvíce?
Nyní zjistíme, jaké jsou počty jednotlivých ras v souboru:
> table(clinical$Race)
asian black hispanic mixed white
10 18 29 42 3 176
Jaké je percentuální zastoupení jednotlivých ras zjistíme pomocí tabulky, kterou uložíme do proměnné a.
> a <- table(clinical$Race)
Nyní převedeme počty na procenta a zaokrouhlíme na jedno desetinné místo:
> b <- a/sum(a)
> is(b)
[1] "table" "oldClass"
> b
asian black hispanic mixed white
0.03597122 0.06474820 0.10431655 0.15107914 0.01079137 0.63309353
Zjistili jsme, že objekt b je tabulka. S tabulkou spojitých hodnot můžeme pracovat jako s čísly, takže ji vynásobíme 100, abychom získali percentuální zastoupení:
> b*100
asian black hispanic mixed white
3.597122 6.474820 10.431655 15.107914 1.079137 63.309353
Nyní zaokrouhlení na jedno desetinné místo:
> round(b*100, 1)
asian black hispanic mixed white
3.6 6.5 10.4 15.1 1.1 63.3
Následuje kontrola kategorické a spojité proměnné: Zkontrolujeme, zda kategorická proměnná ER status správně dělí spojitou proměnnou z imunohistochemie
> boxplot(clinical$ER~clinical$ER_status)
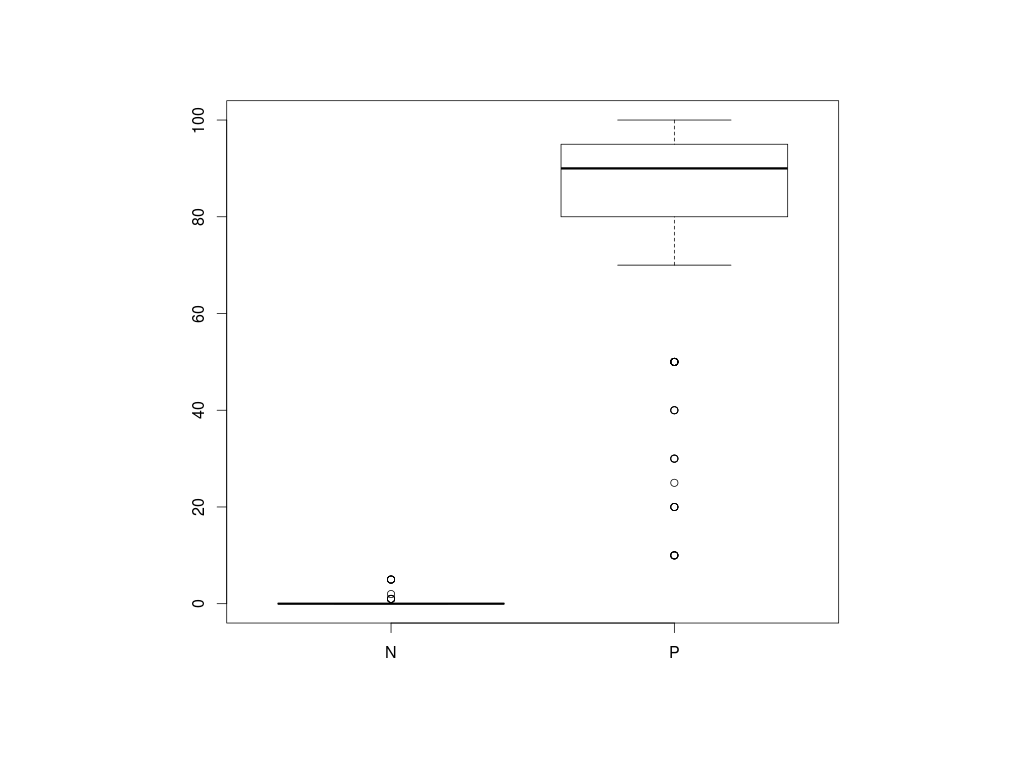
Jaká je hranice pro označení ER pozitivity?
> tapply(clinical$ER, clinical$ER_status, FUN=function(x) quantile(x, na.rm=TRUE))
$N
0% 25% 50% 75% 100%
0 0 0 0 5
$P
0% 25% 50% 75% 100%
10 80 90 95 100
2.Kontrola genových expresních dat
> quantile(X)
0% 25% 50% 75% 100%
2.763678 5.008407 6.078783 7.157690 14.719309
Data jsou v pořádku, měla by být v rozmezí od nuly do patnácti.
