Úprava základních dat
Prvním krokem analýzy dat z TOF MS experimentu je kalibrace. Čas přeletu je přeměněný na škálu m/z pomocí množství kalibračních proteinů se známou m/z hodnotou. Toto se děje ještě v laboratoři. Tak vznikají základní data, které se dále analyzují pro identifikaci píků (proteinů a peptidů) a které vstupují do jednoho ze základních schémat statistické analýzy dat.
Než se tam ale dostanou, musí, podobně jako u mikročipů, proběhnout úprava a normalizace dat, která má dva základní kroky:
- Baseline substraction: Odstranění základního šumu z profilu, například pomocí loess (podobné jako loess normalizace u mikročipů!)
- Normalizace: Tak jako u mikročipů se provádí proto, abychom mohli porovnat spektra mezi vzorky. Odstraňujeme technickou variabilitu (přístrojové chyby, odlišné množství vzorku). Koncentrace proteinu se odhaduje jako plocha pod píkem (Area Under Curve – AUC). Normalizace se děje například pomocí průměrné AUC (TIC – total ion current) podělením AUC celého spektra průměrnou AUC všech spekter.
Obrázky níže ukazují příkladový soubor - výsledek SELDI-TOF jednoho vzorku. Obrázek vpravo již obsahuje červenou křivku, která byla odhadnuta jako baseline, která se odečítá od modré křivky:
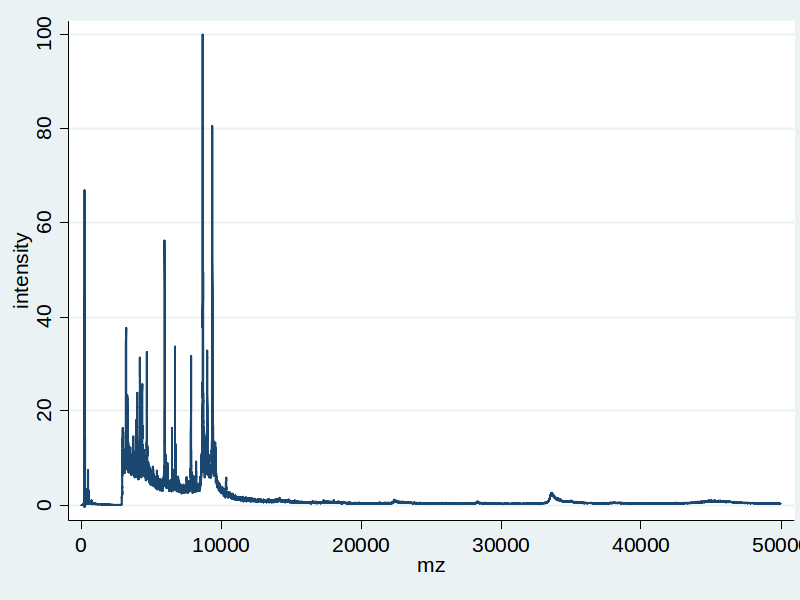
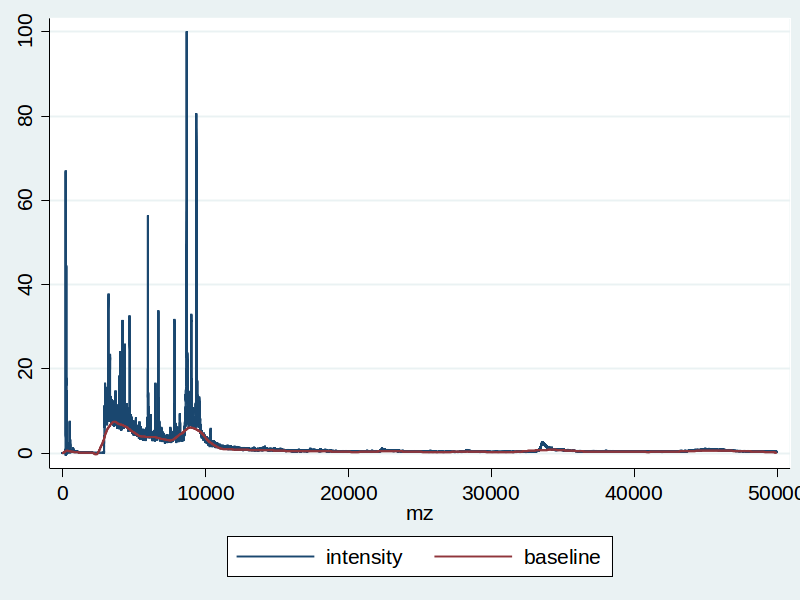
Po odečtení baseline pak upravená data vypadají takhle:
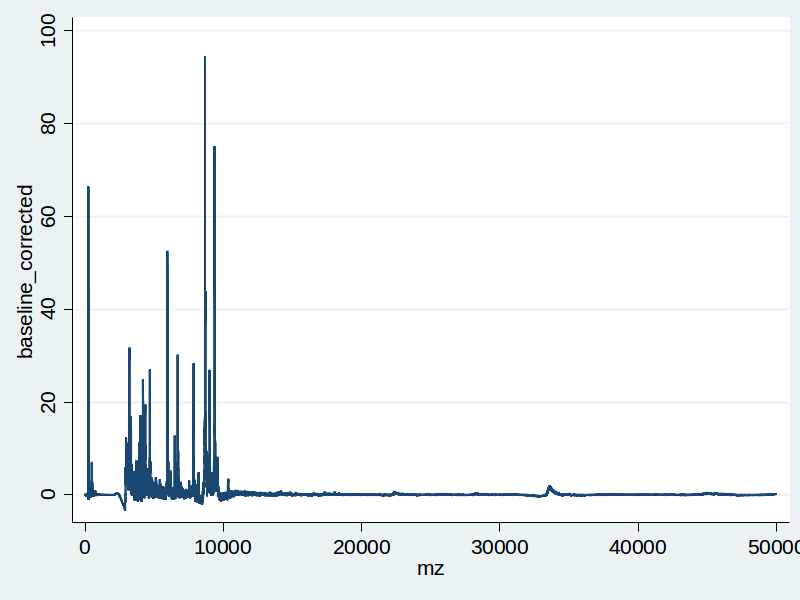
Následuje detekce píků a jejich zarovnání:
Pík reprezentuje peptid nebo protein. Definuje se jako lokální maximum na základě porovnání variability v okolí. Existují nepřesnosti na x (m/z) a y (Intensita) osách. Píky jakéhokoliv spektra mohou být definované jako body, které jsou maximálně +/- N bodů v okolí m/z (podle toho se také detekované píku v některých programech označují jako "first", "second", "estimated", ...). Důležité je brát v úvahu poměr signálu k šumu - jinak řečeno, píky musí překročit nějakou běžnou hranici šumu, aby byli označené za píky. Tato hranice se může měnit, proto je důležité, aby analytik před analýzou dat vědel, jak byli píky identifikovány, nebo ještě lépe, provedl tuto část analýzy sám. V případě porovnání vzorků ze dvou laboratoří se pak totiž zvyšuje technický vliv, který chceme minimalizovat.

Data po zarovnání píků mohou vypadat například takhle:
| Cluster | Group | Norm. Log Intensity | M/Z | Intensity | Norm. Linear Intensity | Type | Mass Dev. |
| 1 | chemoresistentni | 0,581550 | 2392,84 | 3,058176 | 30,578211 | estimated | 0,000007 |
| 1 | chemoresistentni | -0,072123 | 2392,84 | 1,943959 | 12,984676 | estimated | 0,000007 |
| 1 | chemoresistentni | 0,023116 | 2392,84 | 2,076621 | 15,079403 | estimated | 0,000007 |
| 1 | chemoresistentni | 0,16091 | 2392,84 | 2,284742 | 18,36565 | estimated | 0,000007 |
| 1 | chemoresistentni | 0,199591 | 2392,84 | 2,346828 | 19,345988 | estimated | 0,000007 |
| 1 | chemoresistentni | 0,161331 | 2392,82 | 2,285410 | 18,376190 | first | -0,000004 |
V R existuje několik různých balíků pro analýzu dat z hmotnostního spektrometru. Vzpomeneme například balík msProcess (vyřazen z CRAN, ale stále možné ho naistalovat z archivu), msStats, MALDIquant. Principy, které jsme si popsali výše (background substraction a normalizace) jsou stejné pro každý experiment. Co se mění jsou použité metody, podobně jako u mikročipů.
