Analýza dat
1) V analýze budeme muset spojit data genových expresí s klinickými proměnnými. Jde o dvě tabulky, které není třeba spojovat do jedné. Jen je nutné vědět, jak se na ně správně odvolavat.
1a) Musíme zjistit identifikátorový sloupec v klinických datech, který nám propojuje klinická data s daty genových expresí.
Jaké jsou názvy vzorků v genové expresi?
> rownames(X)[1:2]
[1] "GSM505327_19893_AB01778470_17038.CEL.gz" "GSM505328_19893_AB01778504_17045.CEL.gz"
> head(clinical,n=3L)
Sample.name title CEL.file
1 19893_AB01778470_17038 BR_FNA_M157 19893_AB01778470_17038.CEL
2 19893_AB01778504_17045 BR_FNA_M196 19893_AB01778504_17045.CEL
3 19893_AB01778510_17030 BR_FNA_M176 19893_AB01778510_17030.CEL
source.name organism Age Race ER_status
1 Sample ID -- 157, fine-needle aspiration, breast cancer cells homo sapiens 57 white P
2 Sample ID -- 196, fine-needle aspiration, breast cancer cells homo sapiens 69 asian P
3 Sample ID -- 176, fine-needle aspiration, breast cancer cells homo sapiens 77 mixed P
pCR_vs_RD PR_status molecule label description platform
1 RD P total RNA biotin MAQC_Distribution_Status: MAQC_T -- Training GPL96
2 RD P total RNA biotin MAQC_Distribution_Status: MAQC_T -- Training GPL96
3 RD N total RNA biotin MAQC_Distribution_Status: MAQC_T -- Training GPL96
Additional.information Tbefore Nbefore BMNgrd ER HER2.Status Her2.IHC Her2.FISH Histology
1 NA 2 0 2 90 N 2 1.95 IDC
2 NA 2 1 2 90 N 0 1.95 IMC/IDC
3 NA 4 1 2 10 P 2 3.6 IDC
Treatment.Code Treatments.Comments
1 TFAC 98-240 Taxol (80mg/m2 q wk), FAC x4
2 TFAC Taxol 80 mg/m2 weekly x12, FAC
3 TFAC Taxol 80 mg/m2 weekly x12, FAC x1
Sloupec v datech clinical nejbližší názvům řádků v genové expresní matici je třetí s názvem CEL.file.
Stále to ale není to samé, neboť názvy v genové expresní matici mají předponu GSM... a příponu .gz
Odstraníme předponu a příponu názvů řádků matice X, nově vytvořený vektor uložíme do proměnné v
> v<-sapply(rownames(X),FUN=function(x) substring(x, 11, nchar(x)-3))
Nyní porovnáme nový vektor s třetím sloupcem klinického souboru. Pokud názvy sedí, jsou soubory seřazené stejně a můžeme je tak použít.
> match(v, clinical$CEL.file)
[1] 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24
[25] 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48
[49] 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72
[73] 73 74 75 76 77 78 79 80 81 82 83 84 85 86 87 88 89 90 91 92 93 94 95 96
[97] 97 98 99 100 101 102 103 104 105 106 107 108 109 110 111 112 113 114 115 116 117 118 119 120
[121] 121 122 123 124 125 126 127 128 129 130 131 132 133 134 135 136 137 138 139 140 141 142 143 144
[145] 145 146 147 148 149 150 151 152 153 154 155 156 157 158 159 160 161 162 163 164 165 166 167 168
[169] 169 170 171 172 173 174 175 176 177 178 179 180 181 182 183 184 185 186 187 188 189 190 191 192
[193] 193 194 195 196 197 198 199 200 201 202 203 204 205 206 207 208 209 210 211 212 213 214 215 216
[217] 217 218 219 220 221 222 223 224 225 226 227 228 NA 230 231 232 233 234 235 236 237 238 239 240
[241] 241 242 243 244 245 246 247 248 249 250 251 252 253 254 255 256 257 258 259 260 261 262 263 264
[265] 265 266 267 268 269 270 271 272 273 274 275 276 NA 278
> plot(match(v, clinical$CEL.file))
> which(is.na(match(v, clinical$CEL.file)))
[1] 229 277
Zdá se, že vzorky 229 a 277 z genových expresních dat nemají dostupná klinická data.
> clinical[c(229,277),]
Sample.name title CEL.file
229 FL786 642 BR_FNA_M642 FL786 642.CEL
277 FL1141-801(2) BR_FNA_M801 FL1141-801(2).CEL
source.name organism Age Race ER_status
229 Sample ID -- 642, fine-needle aspiration, breast cancer cells homo sapiens 45 hispanic P
277 Sample ID -- 801, fine-needle aspiration, breast cancer cells homo sapiens 45 white P
pCR_vs_RD PR_status molecule label description platform
229 RD P total RNA biotin MAQC_Distribution_Status: MAQC_V -- Validation GPL96
277 RD N total RNA biotin MAQC_Distribution_Status: MAQC_V -- Validation GPL96
Additional.information Tbefore Nbefore BMNgrd ER HER2.Status Her2.IHC Her2.FISH Histology
229 NA 4 2 2 95 N 1.09 IDC
277 NA 2 1 2 91 N 1 IDC
Treatment.Code Treatments.Comments
229 TFEC FEC x 4 Taxol x 12
277 TFEC Taxol x 12 FEC x 4
> v[c(229,277)]
GSM505556_FL786_642.CEL.gz GSM505604_FL1141-801_2_.CEL.gz
"FL786_642.CEL" "FL1141-801_2_.CEL"
> clinical[c(229,277),3]
[1] FL786 642.CEL FL1141-801(2).CEL
278 Levels: FL1141-801(2).CEL FL398-PERU53.CEL FL412-PERU55.CEL FL454-713.CEL ... 29539_AB01833935_35648.CEL
Výsledky nám napovídají, že tomu tak není. Jen se jejich názvy trochu liší.
Obecně se ale zdá, že řádky genových expresních dat i klinických dat si navzájem odpovídají.
2) Vykreslíme si expresi genu ESR1 jako boxplot ve skupině ER+ a ER-
ER+ a ER- je je kódovaná v proměnné ER_status
> clinical$ER_status
[1] P P P P N N P N N P P P N N N N N N P P P N N N P P P P N N N N N P P P P P P P P N N P N N P P N
[50] N P P N N P P P P N N P P P P P P P P P P P P N N P P P P P P P P P N P N P P N N N N P N P N P N
[99] P P N P P P N P N N P N N N N P P P P N P N P P N P P P N P P P P N N P N P P P N N P P N P P P P
[148] N N P N P P P P P P P P N P N N N N N P N N N N N N P P N N N P N P P P N P P N P N P P P P N P P
[197] P P P N N P P N P N P N N N P P N N P N P N N P N P P P N N N N P P N P P N N N N P N P P P N P N
[246] N N N P N P P N P P N N P P P N P P P N P P P P P P P P P P P P P
Levels: N P
Exprese genu ESR1 se skrývá v matici X:
> head(X[,"ESR1"])
GSM505327_19893_AB01778470_17038.CEL.gz GSM505328_19893_AB01778504_17045.CEL.gz
11.218889 11.732910
GSM505329_19893_AB01778510_17030.CEL.gz GSM505330_19893_AB01779182_17047.CEL.gz
10.507449 11.803497
GSM505331_19893_AB01779189_17018.CEL.gz GSM505333_19893_AB01860198_17037.CEL.gz
6.105230 6.266635
Vykreslení boxplotu:
> boxplot(X[,"ESR1"]~clinical$ER_status)
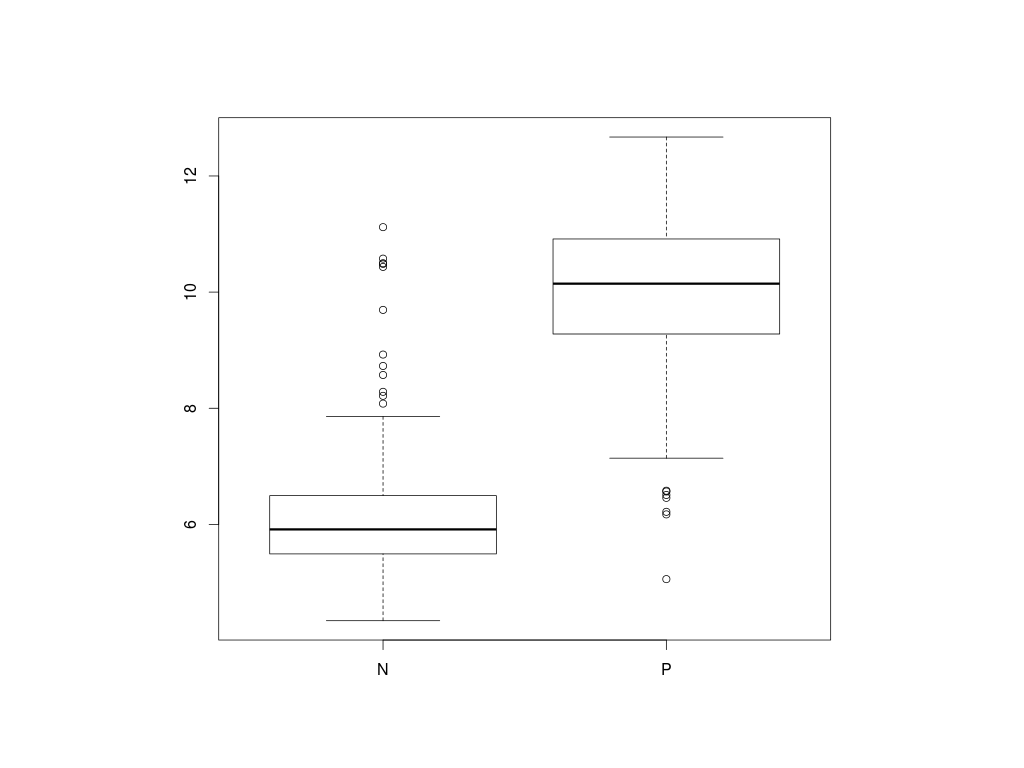
Z grafu vidíme, že exprese genu ESR1 je vyšší ve skupině ER+, kódování jako P. Pomocí t.testu zjistíme, zda je tento rozdíl statisticky významný.
> t.test(X[,"ESR1"]~clinical$ER_status)
Welch Two Sample t-test
data: X[, "ESR1"] by clinical$ER_status
t = -23.0888, df = 245.239, p-value < 2.2e-16
alternative hypothesis: true difference in means is not equal to 0
95 percent confidence interval:
-4.111983 -3.465552
sample estimates:
mean in group N mean in group P
6.251731 10.040498
Rozdíl je statisticky významný. Všimněme si, že jsme použili úplně stejný příkaz jako pro boxplot, jen jsme vyměnili funkci.
3) NAJDEME VŠECHNY ODLIŠNĚ EXPRIMOVANÉ GENY MEZI SKUPINOU ER+ A ER-!
Musíme otestovat gen po genu, ale zautomatizujeme to pomocí funkce apply.
Budeme ukládat jen p-hodnoty a hodnoty statistiky. Proto budeme potřebovat vědět, kde je najdeme. Výsledek t.testu si uložíme do proměnné a, pomocí funkce str() zjistíme její strukturu.
> a<-t.test(X[,"ESR1"]~clinical$ER_status)
> str(a)
List of 9
$ statistic : Named num -23.1
..- attr(*, "names")= chr "t"
$ parameter : Named num 245
..- attr(*, "names")= chr "df"
$ p.value : num 1.93e-63
$ conf.int : atomic [1:2] -4.11 -3.47
..- attr(*, "conf.level")= num 0.95
$ estimate : Named num [1:2] 6.25 10.04
..- attr(*, "names")= chr [1:2] "mean in group N" "mean in group P"
$ null.value : Named num 0
..- attr(*, "names")= chr "difference in means"
$ alternative: chr "two.sided"
$ method : chr "Welch Two Sample t-test"
$ data.name : chr "X[, "ESR1"] by clinical$ER_status"
- attr(*, "class")= chr "htest"
Vidíme, že p-hodnoty jsou uložené pod názvem p.value a statistika pod názvem statistic
> a$p.value
[1] 1.927791e-63
> a$statistic
t
-23.08885
Aplikujeme tedy pomocí funkce apply t.test na sloupce matice X
> p_hodnoty<-apply(X,2,FUN=function(x) t.test(x~clinical$ER_status)$p.value)
> t_statistiky<-apply(X,2,FUN=function(x) t.test(x~clinical$ER_status)$statistic)
a podíváme se na výsledky:
> p_hodnoty[1:5]
A1CF A2BP1 A2M A4GALT A4GNT
0.01068299 0.01305759 0.20258666 0.98709240 0.01059860
> t_statistiky[1:5]
A1CF A2BP1 A2M A4GALT A4GNT
2.57314325 2.50435172 1.27854111 0.01619854 2.57595375
zobrazíme si histogram p-hodnot:
> hist(p_hodnoty)
Kvůli problému mnohonásobného testování ale musíme upravit p-hodnoty, použijeme Benjamini-Hochbergovu kerokci FDR.
> adj_p_hodnoty<-p.adjust(p_hodnoty, method="BH")
Nyní si výsledky uložíme do pěkné datové tabulky:
> ER_vysledky<-data.frame(Gen=names(t_statistiky), Tstat=t_statistiky, pval=p_hodnoty, adjpval=adj_p_hodnoty)
> head(ER_vysledky,n=4L)
Gen Tstat pval adjpval
A1CF A1CF 2.57314325 0.010682987 0.02555406
A2BP1 A2BP1 2.50435172 0.013057592 0.02991322
A2M A2M 1.27854111 0.202586659 0.27012233
A4GALT A4GALT 0.01619854 0.987092397 0.98955304
Nyní tabulku uložíme:
> write.table(ER_vysledky, file="ER_vysledky.txt", row.names=FALSE)
> save(ER_vysledky, file="ER_vysledky.rdata")
Nyní chceme získatinformaci, které geny jsou statisticky významně exprimované mezi skupinami ER+ a ER-:
> ER_vyzn<-which(ER_vysledky$adjpval<0.05)
> head(ER_vyzn)
[1] 1 2 5 6 8 9
A jaké jsou jejich názvy?
> head(ER_vysledky[ER_vyzn,1])
[1] A1CF A2BP1 A4GNT AAAS AADAC AAGAB
Najdeme také geny s vyšší expresí u skupiny ER+:
> ER_vyzn_UP<-which(ER_vysledky$adjpval<0.05 & ER_vysledky$Tstat>0)
> ER_vyzn_UP<-ER_vysledky[which(ER_vysledky$adjpval<0.05 & ER_vysledky$Tstat>0),1]
> ER_vyzn_UP[1:5]
[1] A1CF A2BP1 A4GNT AADAC AARS
Podobně najdeme také významné geny s nižší expresí u skupiny ER-:
> ER_vyzn_DN<-ER_vysledky[which(ER_vysledky$adjpval<0.05 & ER_vysledky$Tstat<0),1]
> ER_vyzn_DN[1:5]
[1] AAAS AAGAB AAMP AASDHPPT ABAT
Chceme najít prvních 100 nejvýznamnějších genů obecně. Musíme tedy geny seřadit podle významnosti, tedy vzestupně na základně adjustované p-hodnoty. Poté můžeme vybrat prvních 100.
> ER_vysl_ord<-ER_vysledky[order(ER_vysledky$adjpval), ]
> head(ER_vysl_ord,n=4L)
Gen Tstat pval adjpval
ESR1 ESR1 -23.08885 1.927791e-63 2.558371e-59
TBC1D9 TBC1D9 -17.44423 6.654162e-44 4.415369e-40
NAT1 NAT1 -16.88252 4.160171e-43 1.840321e-39
CA12 CA12 -16.67447 2.826538e-41 9.377747e-38
> top100<-ER_vysl_ord[1:100,1]
> top100
[1] ESR1 TBC1D9 NAT1 CA12 GATA3 DNAJC12 EVL SLC39A6 IGF1R C6orf211 VAV3
[12] KDM4B MKL2 UGCG DNALI1 P4HTM C5orf30 IL6ST RABEP1 GREB1 AGR2 TFF1
[23] XBP1 IGFBP4 ANXA9 MLPH MAPT TTC39A CELSR1 FOXA1 GFRA1 ABAT APBB2
[34] KIAA0040 SCUBE2 MYB MAGED2 MED13L FAM134B SEMA3F PADI2 TCF7L1 NME3 PBX1
[45] SIAH2 C11orf75 CRNKL1 TFF3 DACH1 ADCY9 G6PC3 CDH3 AKR7A3 ROGDI SCCPDH
[56] ARL3 WWP1 FAM176B SLC19A2 METRN ENPP1 LASS6 LCMT2 SERPINA5 TCEAL1 CIRBP
[67] UBE2I ACADSB FBP1 FUT8 KIAA1467 TRIM29 KIAA0232 IRS1 MREG RHOB ERBB4
[78] BCL2 GOLSYN TPBG TLE3 EGFR IKBKB IFT140 SLC22A5 ZNF688 ABHD2 GAMT
[89] MYO5C MALL RBM47 RAB11FIP1 ANKRA2 RARA ADCY1 KIAA1324 HDGFRP3 KCTD3 FAM171A1
[100] CHST15
> save(top100, file="top100.rdata")
Další úloha k procvičení: Nalezněte geny odlišně exprimované mezi HER2+ a HER2-, uložte je do podobné tabulky a vyexportujte si vektor top100 genů. Použijte jiný název proměnné, abyste si nevymazali stávající výsledek.
Použijeme balík limma. (Lineární model)
> library(limma)
> design<-model.matrix(~0+clinical$ER_status)
> colnames(design)<-c("ERneg","ERpos")
> limma.res<-lmFit(t(X),design)
> cont.matrix<-makeContrasts(ERpos.vs.neg=ERpos-ERneg, levels=design)
Upravíme statistiky pomocí empirické bayesovské metody:
> fit <- contrasts.fit(limma.res, cont.matrix)
> fit2 <- eBayes(fit)
Vykreslíme tabulku top10 nejvýznamnějších genů po adjustaci Benjamin-Hochbergovou procedurou:
> topTable(fit2, adjust="BH")
logFC AveExpr t P.Value adj.P.Val B
ESR1 3.788768 8.486831 23.16433 5.494162e-67 7.291302e-63 140.78993
TBC1D9 2.813598 8.445859 17.83120 5.128835e-48 3.403238e-44 98.09646
CA12 2.595806 8.564933 17.08151 2.738133e-45 1.211259e-41 91.93900
GATA3 2.607238 7.033999 16.55898 2.191524e-43 7.270927e-40 87.64000
NAT1 2.829038 7.920631 15.14380 3.082871e-38 8.182555e-35 76.00428
AGR2 3.088994 9.285334 13.37019 7.460131e-32 1.650057e-28 61.56574
MLPH 1.826468 9.713469 13.31838 1.140716e-31 2.162635e-28 61.14851
EVL 1.606029 7.507148 13.24842 2.022619e-31 3.355273e-28 60.58581
SLC39A6 2.384280 8.926159 13.01214 1.392992e-30 1.889097e-27 58.68995
FOXA1 1.612742 7.165268 13.00948 1.423477e-30 1.889097e-27 58.66868
Všechny geny pak uložíme do proměnné a.
> a<-topTable(fit2, adjust="BH", number=ncol(X))
> head(a,n=4L)
logFC AveExpr t P.Value adj.P.Val B
ESR1 3.788768 8.486831 23.16433 5.494162e-67 7.291302e-63 140.78993
TBC1D9 2.813598 8.445859 17.83120 5.128835e-48 3.403238e-44 98.09646
CA12 2.595806 8.564933 17.08151 2.738133e-45 1.211259e-41 91.93900
GATA3 2.607238 7.033999 16.55898 2.191524e-43 7.270927e-40 87.64000
> hist(a$adj.P.Val)
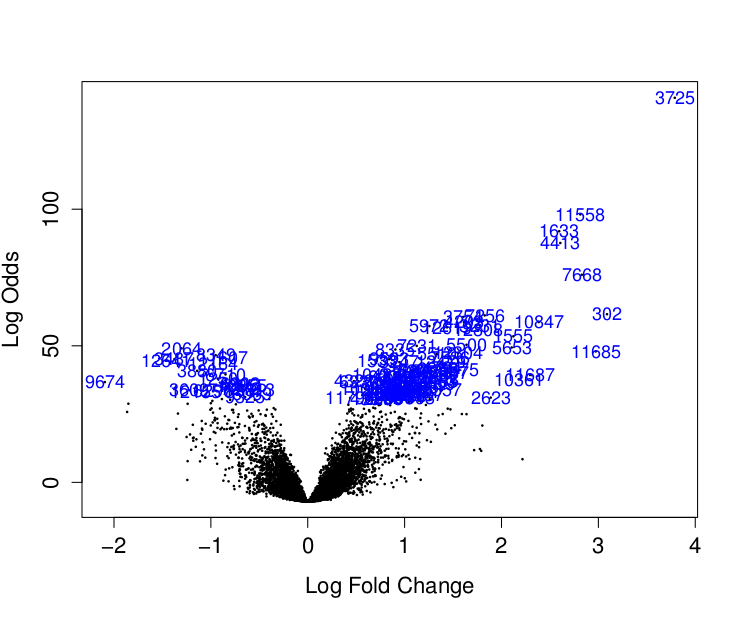
Pro přehlednost ještě vykreslíme volcanoplot:
> volcanoplot(fit2, highlight=100)
Jako další použijeme algoritmus SAM:
> library(samr)
> ER.sam<-c(clinical$ER_status=="P")+1
> data.sam<-list(x=t(X),y=ER.sam, logged2=TRUE, geneid=colnames(X), genenames=colnames(X))
Otestujeme hypotézu, odhadneme s0 a spočítáme očekávané hodnoty d_e. (Výpočet může trvat pár minut)
> samr.obj<-samr(data=data.sam, resp.type="Two class unpaired", nperms=1000)
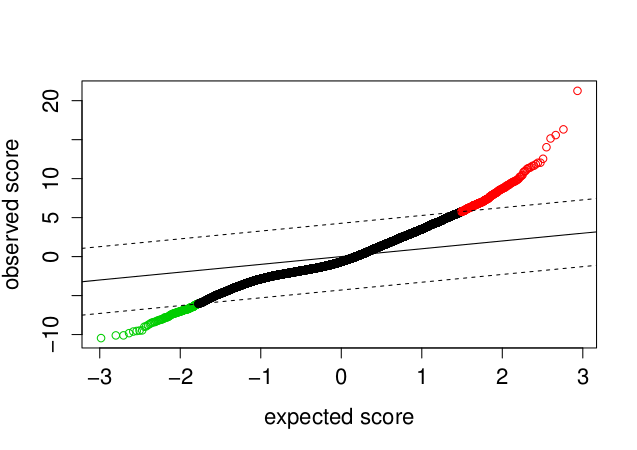
Vykreslíme graf SAM (očekávané versus pozorované hodnoty statistiky)
> samr.plot(samr.obj)
Objekt delta.table je přehled FDR hodnot v závislosti na různém intervalu delta.
> delta.table <- samr.compute.delta.table(samr.obj)
> head(delta.table,n=4L)
delta # med false pos 90th perc false pos # called median FDR 90th perc FDR
[1,] 0.000000000 1920.813 2408.098 11779 0.1630709 0.2044399
[2,] 0.007634704 1901.271 2396.067 11762 0.1616452 0.2037125
[3,] 0.030538818 1850.693 2363.370 11717 0.1579494 0.2017043
[4,] 0.068712340 1764.736 2308.117 11636 0.1516618 0.1983600
cutlo cuthi
[1,] -0.6563521 0.2387363
[2,] -0.6563521 0.2495053
[3,] -0.6563521 0.2797967
[4,] -0.6563521 0.3314694
Výpočet tabulky odlišně exprimovaných genů:
> siggenes.table<-samr.compute.siggenes.table(samr.obj,del=0.4, data=data.sam, delta.table)
> str(siggenes.table)
List of 5
$ genes.up : chr [1:4413, 1:8] "3726" "11559" "1634" "4414" ...
..- attr(*, "dimnames")=List of 2
.. ..$ : NULL
.. ..$ : chr [1:8] "Row" "Gene ID" "Gene Name" "Score(d)" ...
$ genes.lo : chr [1:6652, 1:8] "2065" "12642" "3488" "8350" ...
..- attr(*, "dimnames")=List of 2
.. ..$ : NULL
.. ..$ : chr [1:8] "Row" "Gene ID" "Gene Name" "Score(d)" ...
$ color.ind.for.multi: NULL
$ ngenes.up : int 4413
$ ngenes.lo : int 6652
> head(siggenes.table$genes.up,n=4L)
Row Gene ID Gene Name Score(d) Numerator(r) Denominator(s+s0)
[1,] "3726" "ESR1" "ESR1" "21.2648281846051" "3.78876758517835" "0.178170618275734"
[2,] "11559" "TBC1D9" "TBC1D9" "16.3238237604514" "2.81359768703455" "0.172361434938498"
[3,] "1634" "CA12" "CA12" "15.5907553958805" "2.59580619729516" "0.166496499456405"
[4,] "4414" "GATA3" "GATA3" "15.1565943178548" "2.6072381241063" "0.172020050773209"
Fold Change q-value(%)
[1,] "13.8207843227858" "0"
[2,] "7.03035571417229" "0"
[3,] "6.04526757496136" "0"
[4,] "6.09336061577519" "0"
> head(siggenes.table$genes.lo,n=4L)
Row Gene ID Gene Name Score(d) Numerator(r)
[1,] "2065" "CDH3" "CDH3" "-10.4488321847864" "-1.30239202700949"
[2,] "12642" "VGLL1" "VGLL1" "-10.1031845139517" "-1.4621936349896"
[3,] "3488" "EGFR" "EGFR" "-10.098905543933" "-1.37587531641203"
[4,] "8350" "PADI2" "PADI2" "-9.81242463065464" "-0.950539775347938"
Denominator(s+s0) Fold Change q-value(%)
[1,] "0.124644745362623" "0.405453387989302" "0"
[2,] "0.14472601514606" "0.362940853675632" "0"
[3,] "0.136240042094325" "0.385318853566696" "0"
[4,] "0.0968710396387036" "0.517438829190296" "0"
Regresní strategie:
Vyberte geny, které jsou specifické pro ER status s přihlédnutím na efekt HER2 statusu. Použijeme lineární modely.
Pro jeden gen:
> de1<-lm(X[,2]~ER_status+HER2.Status, data=clinical)
> summary(de1)$coefficients
> de1<-lm(X[,2]~ER_status+HER2.Status, data=clinical)
> summary(de1)$coefficients
Estimate Std. Error t value Pr(>|t|)
(Intercept) 5.27744765 0.02775251 190.161119 7.601085e-294
ER_statusP -0.09405771 0.03306965 -2.844231 4.786213e-03
HER2.StatusP -0.05527367 0.03977918 -1.389512 1.658010e-01
Pro všechny geny: vybereme první (odhad exprese) a čtvrtý sloupec (p-hodnota):
> de<-apply(X, 2, function(z) (
matrix(summary(lm(z ~ ER_status + HER2.Status, data=clinical))$coefficients[,c(1,4)], ncol=1, byrow=FALSE) )
)
> rownames(de)<-c("b0", "ER+", "HER2+", "b0 pval","pvalER", "pvalHER")
adjustace p-hodnot:
> de.adj<-de
> de.adj<-rbind(de,adj.p.ER = p.adjust(de[5,], method='BH'), adj.p.HER2=p.adjust(de[6,], method='BH'))
> de.adj[1:5,1:5]
A1CF A2BP1 A2M A4GALT A4GNT
b0 6.246676e+00 5.277448e+00 8.221261e+00 5.321982e+00 6.134330e+00
ER+ -1.972660e-01 -9.405771e-02 -2.369461e-01 -9.663785e-03 -1.054237e-01
HER2+ -1.657516e-01 -5.527367e-02 -3.584651e-01 -5.587576e-02 -5.520658e-02
b0 pval 8.936419e-231 7.601085e-294 6.925780e-182 1.706690e-269 2.904206e-295
pvalER 3.442293e-03 4.786213e-03 7.897671e-02 8.137026e-01 5.887716e-03
> de.adj.fin<-as.data.frame(t(de.adj))
> head(de.adj.fin)
b0 ER+ HER2+ b0 pval pvalER pvalHER adj.p.ER
A1CF 6.246676 -0.197265990 -0.165751590 8.936419e-231 0.003442293 0.04022286 0.009028196
A2BP1 5.277448 -0.094057711 -0.055273666 7.601085e-294 0.004786213 0.16580096 0.011861407
A2M 8.221261 -0.236946089 -0.358465132 6.925780e-182 0.078976712 0.02740298 0.117962853
A4GALT 5.321982 -0.009663785 -0.055875757 1.706690e-269 0.813702615 0.25786415 0.850957242
A4GNT 6.134330 -0.105423696 -0.055206584 2.904206e-295 0.005887716 0.22795768 0.014005357
AAAS 7.396319 0.079479859 0.003677249 0.000000e+00 0.005926037 0.91512515 0.014086412
adj.p.HER2
A1CF 0.1408932
A2BP1 0.3345959
A2M 0.1118281
A4GALT 0.4357162
A4GNT 0.4046561
AAAS 0.9504180
ER specifické geny, které však nejsou významné u HER2:
> ERspec<-rownames(de.adj.fin)[which(de.adj.fin$adj.p.ER<0.00000001 & de.adj.fin$adj.p.HER2>0.1)]
Geny specifické pro ER+ i HER2+:
> ER.HER2spec<-rownames(de.adj.fin)[which(de.adj.fin$adj.p.ER<0.001 & de.adj.fin$adj.p.HER2<0.001)]
> de.adj.fin[ER.HER2spec[1:5],]
b0 ER+ HER2+ b0 pval pvalER pvalHER adj.p.ER
AAMP 8.069121 0.1941654 0.2910787 3.558697e-308 2.063574e-05 1.440709e-07 1.193715e-04
ABHD11 7.971709 0.3403005 0.6500164 7.701893e-224 2.085872e-04 7.354819e-09 8.827045e-04
ACOX2 5.560283 0.6818004 0.6728123 6.577722e-142 3.295918e-07 2.436741e-05 3.097742e-06
AGR2 6.973743 3.3450704 1.5937433 2.728181e-109 1.823046e-37 9.093824e-09 4.032273e-34
AKT1 7.480583 0.3889358 0.7672964 4.772925e-233 1.273747e-06 1.534804e-14 1.015840e-05
adj.p.HER2
AAMP 1.505485e-05
ABHD11 1.374730e-06
ACOX2 7.754913e-04
AGR2 1.587949e-06
AKT1 1.273024e-11
