SAM - Significance Analysis of Microarrays
SAM - Significance Analysis of Microarrays
Moderovanou statistiku používá metoda Significance analysis of microarrays (SAM). Statistická významnost statistiky dg je stanovena pomocí permutací původních dat a výpočtem očekávaného skóre (de(g)) v případě, že platí nulová hypotéza.
Gen nebo protein je tak označen za statisticky významný, splňuje-li podmínku |dg - de(g) | > Δ.
Algoritmus popisuje následující obrázek:
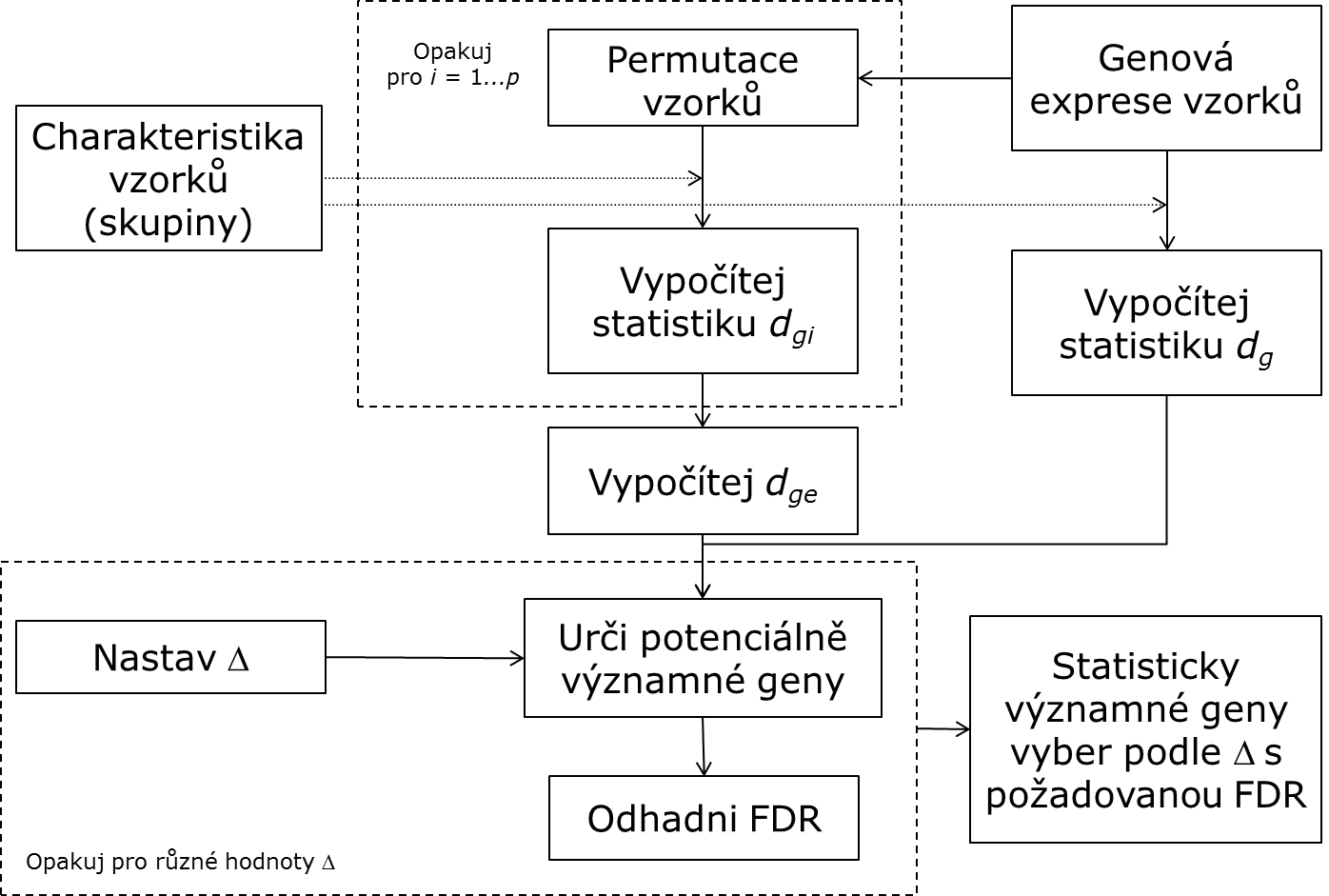
Výpočet (odhad) očekávaného skóre dge probíhá následovně:
1. Pro každou permutaci i:
(i) Pro každý gen g vypočti dig
(ii) Seřaď statistiky všech genů sestupně podle jejich hodnoty
![]()
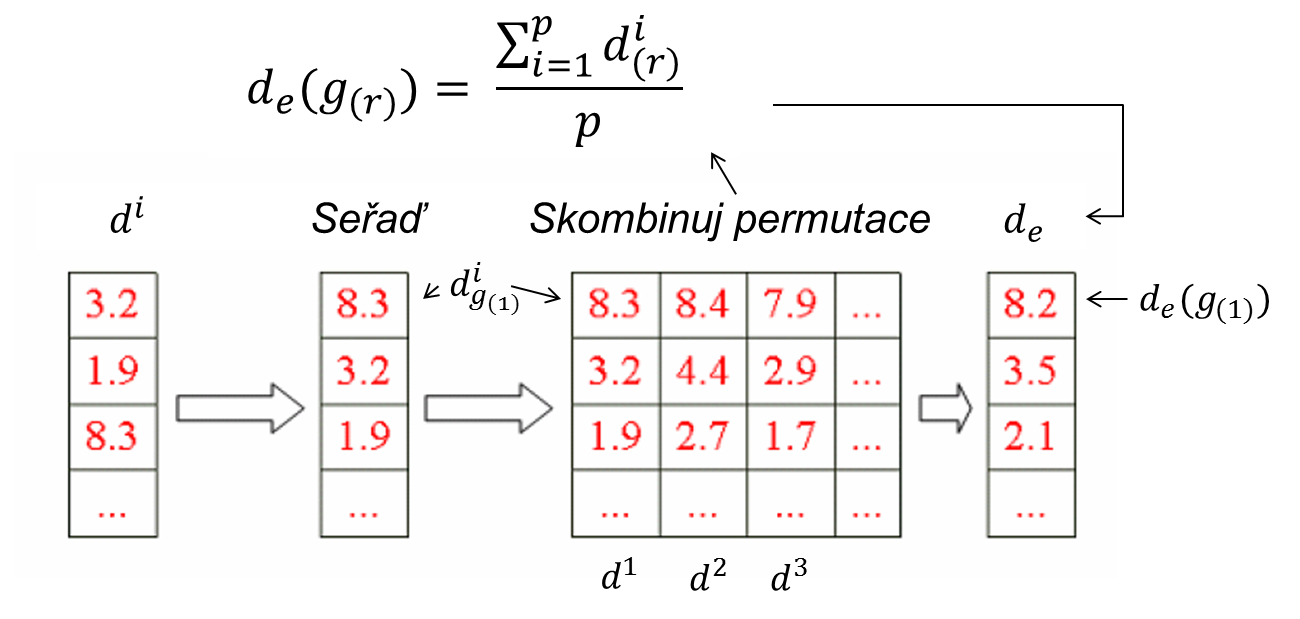
2. Seřaď statistiky původního souboru a zjisti percentil rozložení, který představuje statistika dgi genu g
3. Definuj očekávanou hodnotu statistiky d genu g v původním souboru jako průměr statistik dig z permutovaných souborů na stejné percentilové pozici, jako originální dg jak ilustruje schéma níže:
Máme-li očekávané hodnoty statistiky pro každý gen původního datového souboru, můžeme určit statisticky významné geny.
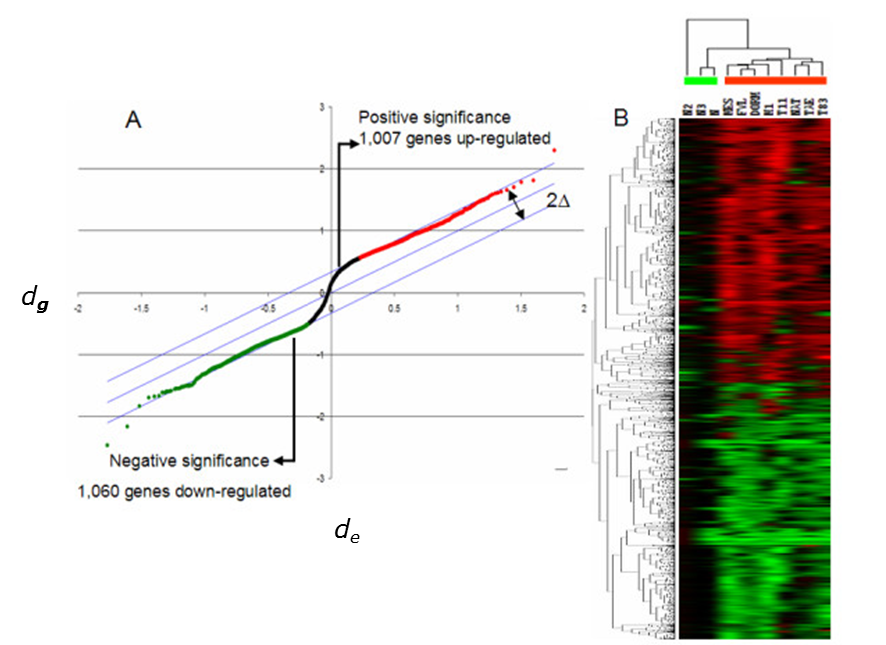
Gen se označí za statisticky významný, pokud platí, že |dg - de(g) | > Δ, jak ukazuje obrázek níže Jak ale vybrat hodnotu Δ ?
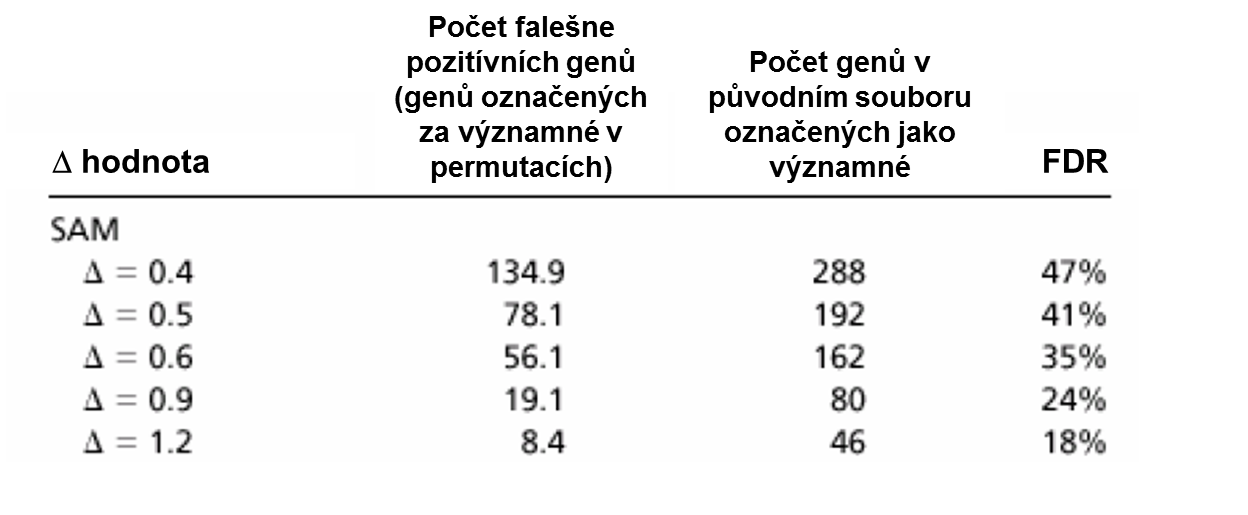
Hodnota Δ se vybere na základě celkové FDR (False Discovery Rate), kterou daná hodnota poskytuje. Pro množinu různých Δ se vypočítá FDR a pak se vybere ta delta, FDR které odpovídá naší dopředu zvolené hladině významnosti. FDR se vypočítá jako:
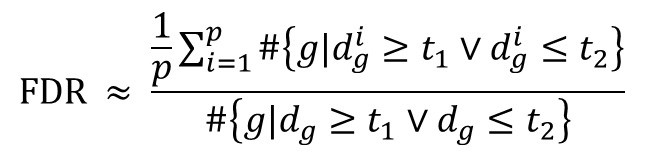 ,
,
kde t1 a t2 jsou dolní a horní hranice statistiky určeny zvolenou Δ. FDR tedy představuje průměrný počet genů, kterých d statistiky v permutacích i=1...p překročili tyto hranice (tedy byly významné), podělený počtem genů v původním souboru, kterých d statistiky překročili tyto hranice. Protože u permutací se očekává, že všechny významné geny jsou falešně pozitivní výsledky, jedná se tedy o odhad podílu falešně pozitivních ze všech označených za pozitivní v původním souboru, což je přesně definice FDR.
Zde bychom chtěli upozornit, že SAM neposkytuje pouze metodu k porovnání dvou skupin. Moderovat můžeme jakoukoliv statistiku a tedy SAM může být aplikován na jakékoliv testování hypotéz. Bioconductor balík samr poskytuje všechny metody, včetně analýzy časových řad:
> source("http://bioconductor.org")
> biocLite("samr")
