Testování hypotéz u genomických a proteomických dat
Testování hypotéz je nepochybně jednou z nejoblíbenějších částí statistiky rozšířené v mnoha vědních oborech. Jde o standardizovanou a dobře interpretovatelnou metodiku, která hraje v analýze genomických a proteomických dat důležitou roli. Zejména když hledáte odlišně exprimované geny/proteiny mezi skupinami. Máte-li nějaké předpoklady a chcete testovat, zda je to pravda, vyžadujete ve skutečnosti dvě výlučná tvrzení, která jsou ve statistice nazývaná hypotézy. Tvrzení, které říká: “Mezi testovanými skupinami není žádný rozdíl” se nazývá nulová hypotéza a tvrzení říkající: “Mezi testovanými skupinami je rozdíl” se nazývá alternativní hypotéza.
Pro seznámení se základy testování hypotéz doporučujeme prostudovat kapitolu učebních textů předmětu Biostatistika pro matematickou biologii, konkrétně kapitoly Úvod do testování hypotéz a Testování hypotéz o kvantitativních proměnných. V dalším textu budeme předpokládat znalosti z těchto kapitol.
Nulovou hypotézou v genomických a proteomických experimentech porovnávajících rozdíl v aktivitě genů nebo abundanci proteinů je v tomto případě tvrzení: "Mezi skupinami není žádný rozdíl v expresi(abundanci) daného genu(proteinu)".
Abychom byli přesnější - testujeme střední hodnoty dvou nezávislých výběrů, kde proměnná je právě gen g a hodnoty jeho exprese u jednotlivých vzorků jsou realizací náhodného výběru o rozsahu n1: x1, x2, …, xn1 (u první z porovnávaných skupin) a na ní nezávislou realizací druhého náhodného výběru o rozsahu n2: y1, y2, …, yn2 (u druhé z porovnávaných skupin).
Všimněte si, že se zde mluví o jediném genu (proteinu) - znamená to, že hypotézu testujeme u každého z genů/proteinů našeho experimentu! To je zdrojem problému, který se nazývá problém (mnoho-)násobného testování hypotéz (klikněte na odkaz pro detailní vysvětlení).
Ve skratce: používáme-li hladinu významnosti 5 % pro rozhodnutí o zamítnutí hypotézy (tzn. že exprese genu je odlišne exprimována mezi skupinami), testujeme-li 10 000 genů, tak 500 z nich (5 %) bude falešne pozitivních. Existují různé druhy korekce na mnohonásobné testování, v analýze genomických a proteomických dat se používají zejména:
- korekce typu FWER (family-wise error rate) - kontroluje pravděpodobnost alespoň jedné chyby prvního druhu - falošně pozitivního (FP) výsledku FWER=PR(FP>0) - patří sem napr. Bonferonniho korekce
- korekce typu FDR (false-discovery rate) - kontroluje očekávaný podíl falešně pozitivních (FP) výsledků mezi zamítnutými hypotézami (Z) FDR=E[FP/Z]- patří sem napr. Benjamini-Hochbergova korekce
FWER je mnohem striktnější a používá se v případě, že chceme, aby všechny vybrané geny/proteiny byly skutečně odlišné mezi skupinami, i za cenu toho, že některé skutečné označíme za falešně pozitivní. FDR je méně striktní a naopak se používá když chceme objevit co nejvíce skutečně pozitivních výsledků, i za cenu falešné pozitivity - to bývá v případě, že tyto geny slouží pro další analýzy jako například analýza genových sad.
Moderovaná T-statistika
U mikročipových dat nelze jednoduše aplikovat klasické statistické testy - zejména Studentův T-test, protože neplatí předpoklad homogenity rozptylů. Je známo, že geny s nízkou hodnotou exprese vykazují mnohem menší variabilitu než geny s vyššími hodnotami exprese, což není pouze reflexe biologie, ale také důsledek technických omezení (jen těžko lze oddělit šum od genu s nízkou expresí). Následkem je vyšší podíl statisticky významných rozdílů v genové expresi právě u genů s nízkou expresí v experimentu, i když tyto rozdíly nejsou biologicky relevantní.
Statistiky genů s nízkou expresí a s vysokou expresí se tak nedají porovnat. V analýze těchto dat se proto používa moderovaná T-statistika, která je definována jako
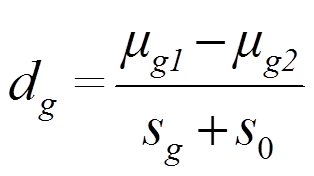
kde![]() a
a ![]() jsou střední hodnoty rozdělení genové exprese skupiny 1 a skupiny 2, sg je směrodatná odchylka. s0 je konstanta, o kterou se zvyšuje variabilita. Tato konstanta musí být zvolena tak, aby nezvyšovala variabilitu u vysoce exprimovaných genů.
jsou střední hodnoty rozdělení genové exprese skupiny 1 a skupiny 2, sg je směrodatná odchylka. s0 je konstanta, o kterou se zvyšuje variabilita. Tato konstanta musí být zvolena tak, aby nezvyšovala variabilitu u vysoce exprimovaných genů.
My si dále představíme dvě metody, které používají moderovanou statistiku: SAM a limma.
