Příklady k procvičení
Nyní si vyzkoušíte své znalosti z této kapitoly na reálném příkladu analýzy genové exprese z cDNA mikročipů.
Cílem experimentu bylo zjistit, co stojí za vývojem Alzheimerovy nemoci a to porovnáním exprese genů mikrocév mozku zdravých jedinců a lidí s Alzheimerovou nemocí (Alzheimer Disease - AD). V experimentu byla izolována RNA z mikrocév mozku u dvou výše uvedených skupin a každý vzorek pak byl hybridizován na cDNA mikročip firmy Agilent.
Data použita v tomto experimentu jsou k dispozici zde http://www.ebi.ac.uk/arrayexpress/experiments/E-GEOD-45596/
My jsme data pro naše účely zjednodušili a pro cvičení bude nutno tyto data stáhnout zde. Zip soubor pak prosím rozbalte do pracovního adresáře. V tomto příkladu budeme pracovat v softwéru R - pro statistické zpracování dat. Pro obeznámení s tímto programem doporučujeme pročíst si výukový text Analýza dat v R.
K načtení základních dat do objektu marray slouží několik funkcí. Nejdříve načtěte balík marray, který obsahuje funkce pro analýzu cDNA dat.
> library(marray)
Následně pomocí funkcí read.marrayLayout a read.marrayInfo načtěte informaci o rozložení spotů na mikročipu a dále o jednotlivých vzorcích:
> brain.layout <- read.marrayLayout(ngr=1, ngc=1, nsr=532, nsc=85)
> brain.targets <- read.marrayInfo(file.path("SampleInfo.txt"), labels=29)
Pomocí funkce maInfo() získáme informace o jménech vzorků, které později použijeme pro zjednodušení názvů sloupců matice exprese:
> jmena<-as.character(maInfo(brain.targets)$'Source Name')
Pomocí funkce read.Agilent() načteme základní data z adresáře "/raw" a vytvoríme objekt třídy marrayRaw, se kterým se bude následně pracovat.
> mraw <- read.Agilent(fnames=dir(path="./raw", pattern=".txt", full.names=T), layout = brain.layout, targets = brain.targets)
Zjednodušíme jména sloupců pomocí vektoru jmena vytvořeného výše:
> colnames(maGf(mraw))<- colnames(maRf(mraw))<- colnames(maRb(mraw))<- colnames(maGb(mraw))<-jmena
> maLabels(maTargets(mraw))<-jmena
Na tomto souboru teď s pomocí funkcí, které jsme si ukázali v této kapitole na příkladu swirl proveďte:
1. Analýzu systematických odchylek u každého čipu
> Gcol <- maPalette(low = "white", high = "green", k = 50)
> Rcol <- maPalette(low = "white", high = "red", k = 50)
> RGcol <- maPalette(low = "green", high = "red", k = 50)
Vykreslíme si heatmapy prvního mikročipu s pomocí funkce maImage
> maImage(mraw[,1], x = "maRb") # vykreslíme pozadí červeného kanálu
> maImage(mraw[,1], x = "maGb") # vykreslíme pozadí zeleného kanálu
> maImage(mraw[,1], x = "maM") # vykreslíme poměr intensit spotů obou kanálů (M hodnoty)
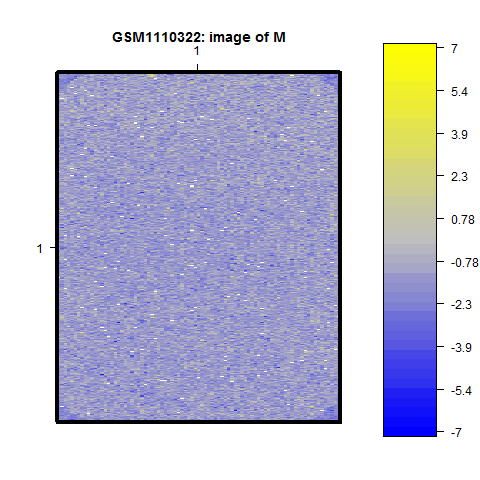
Všimněte si tmavších rohových oblastí na posledním obrázku. To jsou spoty, které jsou na čipu označeny jako "Dark Corners".
S použitím for cyklu a funkce png() můžeme grafy vykreslit automaticky pro všechny čipy a zároveň je uložit do našeho pracovního adresáře
> for (i in c(1:ncol(mraw)))
{
png(paste("imagePlot_",jmena[i],"_maRb.png", sep=""))
maImage(mraw[,i], x = "maRb") # vykreslíme pozadí červeného kanálu
dev.off()
png(paste("imagePlot_",jmena[i],"_maGb.png", sep=""))
maImage(mraw[,i], x = "maGb") # vykreslíme pozadí zeleného kanálu
dev.off()
png(paste("imagePlot_",jmena[i],"_maM.png", sep=""))
maImage(mraw[,i], x = "maM") # vykreslíme poměr intensit spotů obou kanálů (M hodnoty)
dev.off()
}
Inspekcí obrázků zjistíme, že na žádném se nenachází výrazný prostorový efekt.
Určitě jste si všimli, že u některé z obrázků zobrazující pozadí spotu, jako například pozadí červeného kanálu čipu GSM1110312 jsou téměř bílé, s výjimkou několika pixelů.
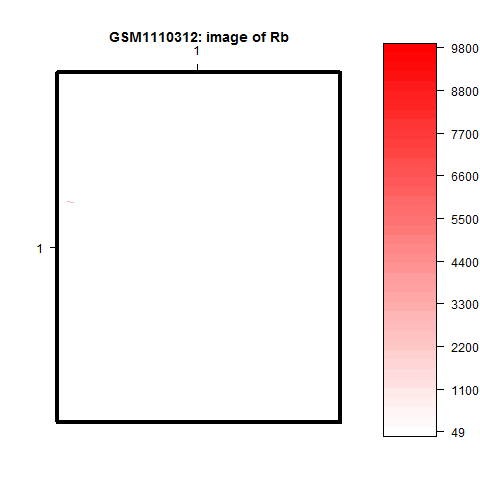
Zde je to způsobeno tím, že několik pixelů má extrémní hodnoty intensity (všimněte si škálu od 49 po 9800!, zatímco u většiny obrázků pozadí se hodnoty pohybují max do 500). Zřejmě u některého spotu proběhl odhad pozadí nesprávně a dostal se tam signál spotu. V případě, že pro normalizaci nebudeme používat korekci na pozadí, nemusíme tuto informaci brát do úvahy. V případě, že ano, musíme zvolit některou globální metodu odhadu pozadí.
Nyní provedeme kontrolu efektu barviva v jednotlivých kanálech. V grafu chceme rozlišit kontrolní sondy a zároveň také spoty, které byly saturovány. Protože informace o saturaci v objektu mraw není, musíme ji načíst ze základních souborů. Vytvoříme si tedy pomocí for-cyklu dvě matice - saturace zeleného a saturace červeného kanálu.
> gIsSaturated <- c()
> rIsSaturated <- c()
> for (i in dir(path="./raw", pattern=".txt", full.names=T))
{
temp <- read.csv(i, header=T, sep="t")
gIsSaturated <- cbind(gIsSaturated, temp$gIsSaturated)
rIsSaturated <- cbind(rIsSaturated, temp$rIsSaturated)
}
> colnames(gIsSaturated)<-jmena
> colnames(rIsSaturated)<-jmena
Jednoduše pomocí základní funkce plot a dvou funkcí, kterými z marrayRaw objektu extrahujeme intensity spotů červeného a zeleného kanálu: vykreslíme graf intensit červeného vs zeleného kanálu:
> R = maRf(mraw)
> G = maGf(mraw)
> for (j in c(1:ncol(mraw)))
{
png(paste("Cy3Cy5_",jmena[j],".png", sep=""), width=2*480)
par(mfrow=c(1,2))
plot(G[,j],R[,j], col=c("blue","red","black")[as.numeric(maControls(mraw))], pch=19, xlab="Green channel", ylab="Red channel", las=1)
abline(a=0, b=1, col="grey", lty=2)
legend("topleft", pch=19, col=c("blue","red","black"), legend=c("neg. kontrola", "pos kontrola", "sonda"))
plot(G[,j], R[,j], col=as.character(c(gIsSaturated[,j]+rIsSaturated[,j])+1), pch=19, xlab="Green channel", ylab="Red channel", las=1)
abline(a=0, b=1, col="grey", lty=2)
legend("topleft", pch=19, col=c("black","red","green"), legend=c("OK", "saturácia Cy3 a Cy5", "saturácia Cy3 alebo Cy5"))
dev.off()
}
Příklad tohoto obrázku:
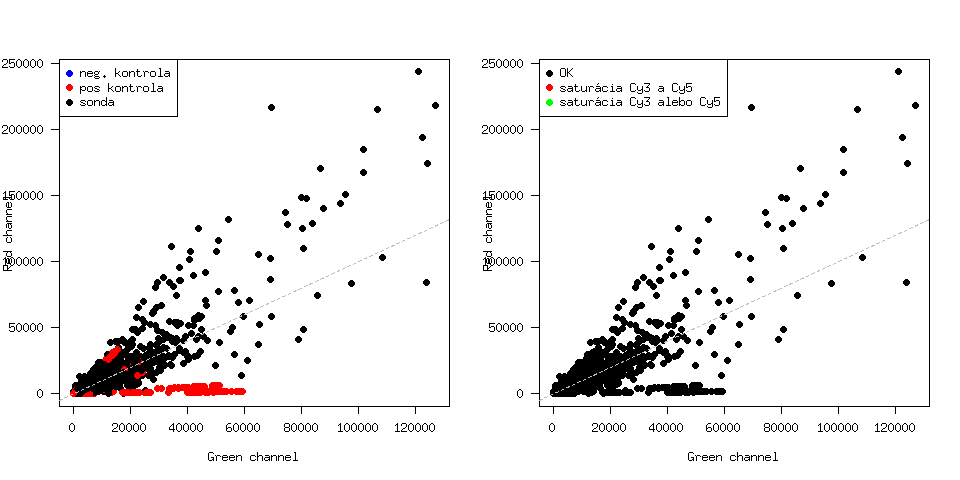
Ani jeden ze spotů není saturován (na obrázku vpravo nejsou žádné barevné body). Nezdá sa, že by se v datech vyskytoval lineární efekt barviva, pro jistotu ješte vykreslíme MA grafy:
Funkce plot aplikována přímo na objekt třídy marrayRaw vykreslí MA graf, s odhadem křivek podle jednotlivých print-tipů
> plot(mraw[,2])
> plot(mraw[,2], col=c("blue","red","black")[as.numeric(maControls(mraw))])
je ekvivalent použití funkce
> maPlot(mraw[,2], col=c("blue","red","black")[as.numeric(maControls(mraw))])
]
2. Normalizaci uvnitř čipu
Aplikujme centrování mediánem na M hodnoty prvního mikročipu z příkladu a zkontrolujme, jak se normalizace (ne)poprala s nelineárními efekty:
> par(mfrow=c(2,1))
> plot(mraw[,1], col=c("blue","red","black")[as.numeric(maControls(mraw))])
> mraw.norm <- maNormMain(mraw[,1], f.loc = list(maNormMed(x=NULL,y="maM")))
> plot(mraw.norm[,1], col=c("blue","red","black")[as.numeric(maControls(mraw))])
Prohlídněte si MA plot dat nenormalizovaných a normalizovaných pomocí centrování mediánem - jaký je rozdíl?
A teď aplikujme normalizaci pomocí loess a znovu vykresleme MA plot:
> par(mfrow=c(2,1))
> plot(mraw[,1], col=c("blue","red","black")[as.numeric(maControls(mraw))])
> mraw.norm.loess <- maNormMain(mraw[,1], f.loc = list(maNormLoess()))
> plot(mraw.norm.loess, col=c("blue","red","black")[as.numeric(maControls(mraw))])
Jaký je výsledek normalizace teď? Která normalizace je lepší?
]
3. Normalizaci mezi čipy
Funkce maBoxplot vykreslí krabicové grafy
> maBoxplot(mraw)
Provedeme normalizaci pomocí loess a následně škálovou normalizaci mezi čipy a znovu vykreslíme krabicové grafy.
> par(mfrow=c(2,1), mar=c(1,3,0,0))
> mraw.norm <- maNormMain(mraw)
> maBoxplot(mraw.norm)
> mraw.norm.scale = maNormScale(mraw.norm)
> maBoxplot(mraw.norm.scale)
]
