Supermatice
Vstupní data pro rekonstrukci stromu druhů ze supermatic jsou alignmenty jednotlivých lokusů seřazené za sebou tak, aby jednotlivé sekvence odpovídaly taxonům. Taková sekvence se nazývá spojená sekvence (concatenated). Chybějící sekvence lokusů u některých taxonů bývají v supermatici značené jako chybějící nebo nerozlišené báze (obr.2).
Otázka: Proč není vhodné nejdřív spojit sekvence všech lokusů pro každý druh a až pak je alignovat?
Takový přístup sebou nese riziko, že počátky a konce lokusů nebudou seřazeny podle homologie v rámci lokusu, ale na základě náhodné podobnosti se sousedící sekvencí. Navíc, pokud nejsou k dispozici sekvence všech lokusů pro všechny taxony, spojená sekvence určitě nebude správně zalignovaná s dlouhou delecí v místě chybějícího lokusu.
Kromě nutnosti správně alignovat i spojené sekvence, u kterých chybí některý lokus, je dobré spojovat až alignmenty i při mezidruhové analýze. V genomu se jednotlivé lokusy můžou vyskytovat v různém pořadí, anebo geny můžou být na reverzně komplementárních řetězcích DNA.
Sestavení supermatice je často motivované snahou o využití sekvencí z genetických databází. Spojením sekvencí z několika jedinců vzniká chimerická sekvence (obr. 3) s rizikem, že zveřejněné sekvence mohou patřit k jinému taxonu. Problém se dotýká taxonomických skupin, u kterých je podezření na dosud neodhalenou kryptickou diverzitu.
Spojené a hlavně chimerické sekvence často neobsahují všechny lokusy pro všechny taxony. Sekvence DNA je ale natolik informativní, že umožňuje spolehlivě rekonstruovat fylogenezi i když je procento chybějících dat vysoké, běžně nad 60%.
Analýza supermatice má specifika při definování substitučního modelu. U alignmentu z mnoha genů je pravděpodobné, že jednotlivé geny anebo skupiny genů bude optimálně modelovat jiný substituční model. Supermatice by měla být rozdělena do particí podle volby substitučního modelu. Následná analýza jak metodou ML tak i BI probíhá obdobně jako u genových alignmentů.

Obr. 2: Ukázka supermatice s vysokým procentem chybějících dat. Každý řádek představuje spojenou sekvenci pro jeden taxon. Široké, černé bloky jsou úseky, pro které je k dispozici sekvence DNA. Úzké, černé bloky jsou sekvence s nejasnými rezidui (DNA ambiguity). Bílé bloky jsou indely a chybějící části nebo celé lokusy. Vertikální čáry oddělují jednotlivé alignmenty lokusů. Uvedená supermatice má celkovou délku 9065 bp, skládá se z osmi lokusů, 21 taxonů a obsahuje 67% chybějících dat.
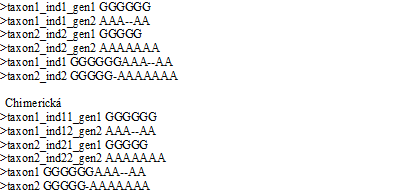
Obr. 3: Spojená sekvence pro jednoho jedince a pro chimerickou sekvenci, kde jednotlivé lokusy patří ke zkoumanému druhu, ale pocházejí z různých jedinců.
