Predikce eukaryotických genů
V eukaryotickém genomu tvoří pojmenované geny často méně než 2% celé genomické sekvence. Nalézt takové geny je proto komplikovanější než u prokaryot a vyhledávání ORF není dostatečné.
Otázka: Proč nestačí vyhledat u eukaryot ORF a následně vytřídit vhodné kandidáty na základě promoterových sekvencí jako u prokaryot?
Skutečných ORF by bylo velice málo v porovnaní s možnými kombinacemi start a stop kodonů. Délka genů je mnohem variabilnější než u prokaryot a většina eukaryotických genů obsahuje introny, které můžou posouvat ORF při přímém čtení.
Struktura genů u eukaryot je komplexnější a výjimkou nejsou ani geny, které mají délku řádově v 105 bp. Velmi dlouhé geny budou rozděleny na exony a introny, přičemž introny budou výrazně delší než exony (Obr. 3). Pro účely predikce genů je dobré si uvědomit, že eukaryotický gen se přepisuje do mRNA včetně nepřekládaných oblastí na 5‘ a 3‘ koncích (UTR) a intronů. UTR ale nejsou predikovány a přesný rozsah přepisovaného genu není z predikcí patrný. Predikují se kódující sekvence od start po stop kodon a určuje se lokace a počet intronů a pro zjištění UTR oblastí je potřebné osekvenovat mRNA molekulu. Zároveň tyto experimenty poskytují důkazy o expresy daného predikovaného genu, zlepšují určení intronů a případně poskytují informace i o alternativním sestřihu u daného genu.
Predikce eukaryotických genů je založena na vyhledávání regulačních sekvencí pro transkripci. Příklady takových sekvencí jsou:
- TATA box = TATAAA (podobné Pribnowově sekvenci u prokaryot - TATAAT)
- iniciační element = YYANWYY
- GC box = GGGCG
- CAAT box = GGCCAATCT
- BRE (z angl. B recognition element) = SSRCGCC
Symboly N, S, R, Y a W v sekvencích představují kódy nejistoty v DNA (Tab. Contig.1). Jak konkrétně bude daná sekvence vypadat u toho kterého genu a organizmu může být značně specifické a/nebo variabilní. Na jejich vyhledávání je proto znovu potřeba sofistikovanějších metod, jako např. skryté markovovy modely, neurální sítě nebo diskriminace vzoru. Všechny tyto metody mají jedno zásadní omezení. Jsou parametrizované a je tedy nutné před samotnou predikcí určit hodnoty pro dané parametry – „natrénovat“ danou metodu na známých datech.
Po identifikaci oblasti DNA sekvence, která by mohla kódovat gen u eukaryot, je dále nutné zjistit, kde konkrétně jsou kódující úseky, čili exony. Taková predikce musí pro úspěšnou translaci funkčního proteinu splňovat několik vlastností. První exon začíná start kodonem a poslední končí stop kodonem. Introny nesmí rozdělovat start a stop kodony, ale můžou rozdělit vnitřní kodony. Po sestřihu mRNA musí sekvence představovat souvislý ORF.
Predikce lokalizace intronů probíhá kombinací sensorů obsahu a signálu. Signál je v tomto případě obvyklá donorová sekvence GT na začátku intronu a akceptorová AG na jeho konci. Sensor signálu představuje očekávaná frekvence nukleotidů v okolí predikovaného začátku a konce intronu (Obr. 4).
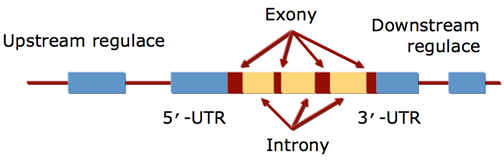
Obr. 3: Struktura genu u eukaryotického organizmu. UTR – nepřekládaný úsek (untranslated region).
Obr. 4: Grafické znázornění frekvence nukleotidů v okolí rozhraní exonů a intronů u pěti lidských genů pro interleukiny a korespondující váhová matice. Intron začíná na pozici 7.
