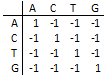Lokální alignment
Lokální alignment hledá maximální podobnost sekvencí na krátkém úseku sekvencí a je založený na Smith-Watermanově algoritmu. Jedná se o algoritmus dynamického programování, který rozdělí komplexní problém na sérii malých úkolů. V případě alignmentu to znamená, že namísto alignování celé sekvence, porovnáváme shodu nebo rozdíl u dvojic nukleotidových pozic (obr.2). Porovnávání je pak skórováno podle zvoleného schématu. Na uvedeném příkladu je skóre za shodu (match) +1 a za rozdíl (mismatch) –1.
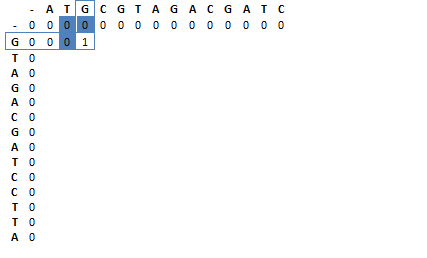
- Sekvence 1 se vypíše do názvů sloupců tabulky, sekvence 2 do názvů řádků. Smith-Watermanův algoritmus nepovoluje negativní hodnoty při skórování a ty jsou vynulovány. V prvním řádku a sloupci jsou tedy nuly.
- První pozice obou sekvencí se porovná a skóre se připočítá k nejvyšší hodnotě v buňkách nad, vlevo a vlevo nahoře v diagonále od doplňované buňky.
- Obdobným způsobem se vyplní celá tabulka (obr. 3).
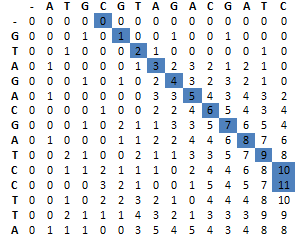
- Ve vyplněné tabulce se nalezne maximální hodnota, která představuje skóre alignmentu (Blast).
- Od té se postupuje zpět po nejvyšších hodnotách v sousedících buňkách nad, vlevo a vlevo nahoře. Pokud jsou skóre v hodnocených buňkách stejné, upřednostňuje se směr po diagonále.
- Zpětné sledování (traceback) končí, když algoritmus narazí na nulu.
- Sekvence se zalignují ve směru zpětného sledování. Směr po diagonále znamená seřazení nukleotidových pozic, směr nahoru zařazení mezery (gap) do sekvence 1, směr doleva zařazení mezery do sekvence 2.
|
A |
T |
G |
C |
G |
T |
A |
G |
A |
C |
G |
A |
T |
- |
C |
- |
- |
- |
|
- |
- |
- |
- |
G |
T |
A |
G |
A |
C |
G |
A |
T |
C |
C |
T |
T |
A |
Obr. 3: Lokální alignment sekvencí z obr. 1. Modrá pole představují spětné sledování (traceback, nahoře) nejvyšších hodnot skóre alignmentu, podle kterých se sekvence zarovnávají (dole).
Při porovnání alignmentu sestaveného ručně na obr. 1 a vypočítaného pomocí Smith-Watermanova algoritmu na obr. 3 vidíme rozdíl v pozici mezery na 3‘ konci sekvence 1. Optimalizace alignmentů je intenzivně řešená problematika a v současnosti program s nejvyšším procentem správně alignovaných párů sekvencí je MAFFT.
Princip dynamického programování je dobré pochopit kvůli pochopení podstaty skóre alignmentu, které je důležité při hodnocení výsledků z blastu (Blast). Skóre alignmentu orientačně informuje nejenom o míře shody mezi sekvencemi, ale i o délce sekvence, ve které se shoda hodnotí.
Hodnoty odměn a penalt za shodu a rozdíl nukleotidových bází bývají obyčejně stejné pro všechny záměny. Skórovací matice má jenom dvě hodnoty (obr. 4). Složitější skórovací matice se používají k alignování sekvencí aminokyselin. Zde je důležitý fakt, že některé aminokyseliny jsou si chemicky podobné a tedy jejich záměna nemusí výrazně ovlivňovat funkčnost proteinu. Skórovací matice pro aminokyseliny mají empiricky určené hodnoty. Mezi nejznámější patří BLOSUM62, JTT nebo WAG.